











 nueva página del texto (beta)
nueva página del texto (beta) Inglés (pdf)
Inglés (pdf)
 Artículo en XML
Artículo en XML Referencias del artículo
Referencias del artículo
 Enviar artículo por email
Enviar artículo por email Citado por SciELO
Citado por SciELO  Similares en
SciELO
Similares en
SciELO 
 Permalink
Permalink1. Introduction
Formaldehyde (H2CO) is an important reactive intermediate product in tropospheric hydrocarbon oxidation initiated by the OH radical. Its concentration in the atmosphere is in the range of 1 to 10 ppb [1]. The importance of H2CO molecule originates from its widely use in many industrial manufacturing processes due to the high chemical reactivity and good thermal stability. On the other hand, it is a well-known pollutant that is emitted through incomplete combustion processes [2]. To date, several methods have been developed to detect the H2CO concentrations which have been reviewed comprehensively by Vairavamurthy et al. [3-7]. Since the discovery of fullerenes, numerous studies have been focused on the nanotubes, nanoclusters, nanocones, nanocapsules, nanoribbons, etc [8-16]. BN nanostructures are wide band gap materials, expecting to show special electronic, optical and magnetic properties such as Coulomb blockade and supermagnetism [17]. The properties of BN fullerenes are different with those of carbon fullerenes, from the viewpoint of electronic properties and thermal resistance. Geometries and stability of (BN)n (n = 4-30) nano-cages have been previously studied by various research groups [18-20]. B12N12 cluster was theoretically shown to be more stable with the structure based on decoration of truncated octahedrons in which all B vertices remain equivalent, as well as all N, and was successfully synthesized [18].
Recently, Wu et al. have investigated hydrogenation of a B12N12 molecule by calculations using ab initio molecular orbital theory [21]. We have previously shown [22] that B12N12 is the most stable nanocluster among different X12Y12 (X = Al or B and Y = N or P) cages. Nanostructured materials have been invoked more attention as chemical sensors due to high surface to volume ratio and high electronic sensitivity [23-29]. It has been previously demonstrated that the electronic properties of pristine BN nanotubes and graphene-like sheets are not sensitive toward presence of H2CO gas, and cannot be used as chemical sensors [29, 30]. Here, the interaction between an H2CO molecule and a B12N12 nanocluster is investigated using density functional theory (DFT) calculations to answer this question that whether there is a potential possibility of BN nanoclusters serving as chemical sensor to H2CO.
2. Computational methods
Geometry optimizations, natural bond orbitals (NBO), and density of states (DOS) analyses were performed on a B12N12 nanocluster and different H2CO/B12N12 complexes at the spin unrestricted X3LYP level [31] of theory with 6-31G (d) basis set [32] as implemented in GAMESS suite of program [33]. GaussSum program [34] was used to obtain DOS results. Vibrational frequency calculations were performed using numerical second derivatives, verifying that all the structures are true minima with positive Hessian eigenvalues. The Gibbs free energy change (ΔG) of H2CO adsorption at room temperature and 1 atm pressure is defined as follows:
where G(H2CO/B12N12) is the Gibbs free energy of complex, and G(B12N12) and G(H2CO) are the Gibbs free energies of the pristine B12N12 and H2CO molecule, respectively. Zero-point and basis set superposition error (BSSE) corrections were included in the ΔG calculations. HOMO-LUMO energy gap (Eg) is defined as
where ELUMO and EHOMO are energies of HOMO and LUMO. When we evaluate the properties of the sensor, the shift of the Eg is obtained by:
where Eg1 and Eg2 are, the value of the Eg for clean B12N12 and the formaldehyde adsorbed state, respectively.
3. Results and discussion
The B12N12 (Fig. 1) nanocluster is made of six squares and eight hexagons. There are two types of individual B-N bond among the all 36 B-N bonds; one is shared by two six-membered rings (B66), and another by a four- and a six-membered ring (B64). The B64 bond (average length = 1.48 Å) is somewhat longer than the B66 one (average length = 1.43 Å). NBO population analysis shows a net charge transfer of 0.45 electrons from B to N atom in the surface of the cluster, indicating partly ionic nature of these bonds. Several distinct starting structures were considered for H2CO adsorption on the cluster. For example, the oxygen or a hydrogen atom of H2CO molecule was placed atop a hexagon or a square ring, or the molecule was located close to B64 or B66 bond so that its O and C or H atoms were close to the B and N atoms of these bonds. However, after careful relax optimization of initial structures four different stable structures (local minima) were obtained which are shown in Fig. 2-Fig. 4 (Fig. 3).
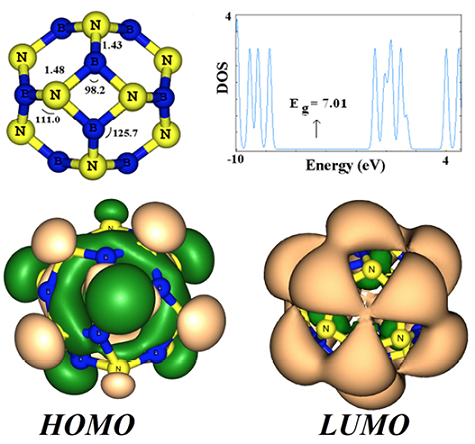
Fig. 1 Structural model, HOMO, LUMO profiles and the electronic density of states (DOS) of the B12N12 cluster. (Bonds are in angstrom and angles are in degree).

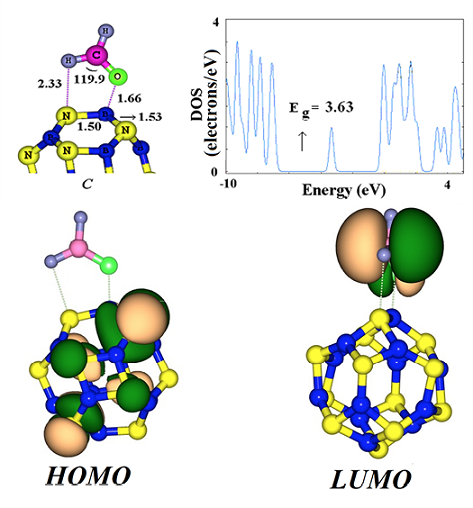
Fig. 3 The optimized structure of H2CO chemisorption on the B12N12 cluster and its DOS plot, HOMO, and LUMO profiles.
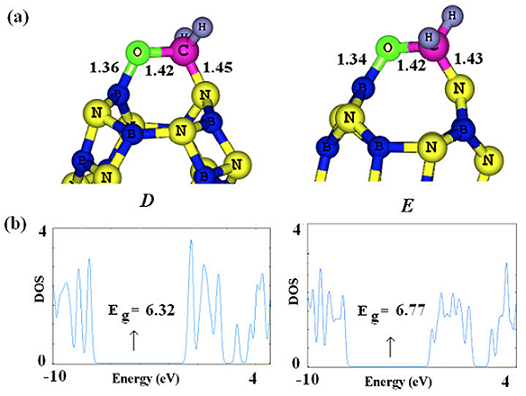
Fig. 4 The optimized structures of chemically functionalized B12N12 cluster with H2CO and their DOSs.
The interactions can be divided into three types: (I) Physisorption, in which H2CO molecule weakly interacts with the cluster through van der Waals forces and less negative ΔG values (configurations A, Fig. 2). (II) Chemisorption, in which the ΔG value is rather more negative than that of the physisorption, and an insignificant change occurs in the geometrical parameters (configuration B, Fig. 3). (III) Chemical functionalization, including very strong interaction that largely deforms the structure of the cluster with bond cleavages and formations (configurations C and D, Fig. 4). The ΔG values, charge transfer, and electronic properties for the all configurations are summarized in Table 1. For the physisorption configuration A, the calculated values of ΔG is about -0.07 eV. Also, the corresponding interaction distances between the N atom of B12N12 cluster and the H atom of H2CO molecule is 3.30 Å. The less negative ΔG values and large interaction distances indicate that the H2CO molecule undergoes a weak physical adsorption due to van der Waals forces. The DOS for this configuration is shown in Fig. 2. It can be found that the DOS of pristine BN cluster (Fig. 1) is slightly affected by the adsorption of H2CO molecule near the Fermi level and ΔEg is negligible. The charge transfer from the H2CO molecule to the cluster is very small (about 0.02 e).
Table 1 Calculated Gibss free energy change (ΔG), HOMO energies (EHOMO), LUMO energies, HOMO-LUMO energy gap (Eg), and Fermi level energy (EF) of formaldehyde adsorbed on the B12N12. Energies are in eV. See Figs. 2, 3 and 4.
| system | ΔG | LUMO | EF | HOMO | Eg | ΔEg (%) | aQT (|e|) |
|---|---|---|---|---|---|---|---|
| cage | - | -1.63 | -5.14 | -8.64 | 7.01 | - | - |
| A | -0.07 | -1.65 | -4.77 | -7.88 | 6.23 | 11 | 0.02 |
| B | -0.59 | -2.77 | -4.59 | -6.4 | 3.63 | 48 | 0.21 |
| C | -2.00 | -1.07 | -4.23 | -7.39 | 6.32 | 10 | -0.04 |
| D | -1.55 | -1.09 | -4.48 | -7.86 | 6.77 | 3 | -0.02 |
a QT is defined as the total NBO charge on the formaldehyde
As shown in Fig. 3, the distances between the H and O atoms of H2CO, and the N and B atoms of the cluster in the configuration B are about 1.66 and 2.33 Å, respectively. The ΔG value is about -0.59 eV with a charge transfer of 0.21 e from the H2CO molecule to the cluster. The adsorption induces a slight structural deformation to both the adsorbed molecule and the B12N12 nanocluster (Fig. 3). Angles of H-C-H and H-C-O of H2CO are changed from 115.2° and 122.3° in their free state to 121.5° and 119.9° in the adsorbed form, and the H2CO-adsorbed B-N bond is pulled outward from the cluster surface with the bond length increasing from 1.43 Å (pristine cluster) to 1.50 Å. In order to explore the sensitivity of B12N12 toward H2CO molecule, we plotted the DOS of this structure and compared it with that of free cluster.
As shown in Fig. 3, the DOSs of the configuration B near the conduction level has an appreciate change compared to that of the pristine cluster, so that a local energy level appears after the adsorption of H2CO molecule. Our frontier molecular analysis shows that in consistent with the energy change the LUMO shifts on the H2CO molecule while the HOMO is still remained on the cluster (Fig. 3). Fig. 1 indicates that in the pristine cluster the HOMO and LUMO is mainly located on the N and B atoms of the cluster, respectively. However, based on the DOS analysis, the Eg value of the cluster decreases from 7.01 to 3.63 eV (48% change) upon the H2CO molecule chemisorption on the BN cluster, which would result in the electrical conductivity change of the cluster. This phenomenon can be interpreted by the following relation [35]:
Where σ is the electrical conductivity, A (electrons/m3K3/2) is a constant, and k is the Boltzmann's constant. According to the equation, smaller Eg at a given temperature leads to the larger electrical conductivity. It has been previously [36] shown this equation is compatible with the experimental results, and larger Eg at a given temperature leads to smaller electrical conductivity.
However, it seems that the cluster can transform the presence of the H2CO directly into an electrical signal, suggesting that, B12N12 nanocluster may be a sensitive gas sensor toward the toxic H2CO molecules. We found that this behavior of the BN nanocluster is in contrast to that of the BNNTs reporting by Zhang et al . [30]. They have theoretically investigated the adsorption of H2CO molecule on the BNNTs, showing that the electronic properties of pristine types of these nanotubes are not affected by the adsorption of H2CO molecule. Recovery of the sensor devices is of great importance. Based on the conventional transition state theory, the recovery time, τ, can be expressed as [36]:
where T is temperature, k is the Boltzmann's constant, and ν0 is the attempt frequency. According to this equation, an increase in the ΔG, will prolong the recovery time in an exponential manner.
As shown in Table 1, the ΔG of H2CO chemisorption is about -0.59 eV which is not too large to prolong the desorption process. Finally, we considered two possible configurations that in which the cluster undergoes a strong chemical functionalization with the H2CO molecule. To this end, the C-O bond of H2CO molecule was horizontally located close to a B-N bond of the cluster, and then a full relax optimization was performed. The functionalized structures are shown in Fig. 4, in which the H2CO molecule strongly adsorbed on the B64 (configuration C) or B66 (configuration D) bonds. Based on the NBO results and geometry analysis, both the B64 and B66 bonds are broken after the adsorption process and two new bonds are formed, namely, C-N and O-B. The configurations C and D have ΔG of -2.00 and -1.55 eV, with rather a small charge transfer about 0.04 and 0.02 e from the cluster to the H2CO molecule, respectively. Geometric parameters such as the increased C-O bond length of H2CO molecule (1.42 Å in configurations C and D compared to 1.20 Å in the isolated H2CO), reduced H-C-H bond angle (109° as compared to 115.2° in the isolated H2CO), and deviation of the hydrogen atoms from the original molecular plane, clearly indicate increasing p character of molecular orbitals on the C and O atoms. The NBO analysis shows that the hybridization of C and O atoms of free formaldehyde is about sp2.11 and sp1.42 which change to sp3.14 and sp2.85 in D configuration, respectively.
As it is shown in Table 1, the released energy during the chemical functionalization of the B64 bond is somewhat larger than that of the B66 bond. It can be interpreted by the fact that the B64 bond is shared by six- and four-membered rings while the B66 by two six-membered ones. Since a four-membered ring is thermodynamically more unstable than a six-membe red one due to high strain, its cleavage is energetically easy in comparison to that of the six-membered ring, therefore, chemical functionalization of the B64 bond is thermodynamically more favorable than that of the B66 one.
The electronic properties of the cluster are not significantly changed upon the formaldehyde adsorption via these configurations. The DOS plots are shown in Fig. 4, indicating that the Eg value of cluster is slightly decreased by 0.69 and 0.24 eV in the C and D configurations, respectively. Despite the thermodynamic feasibility of the chemical functionalization, the process does not occur in room temperature due to obvious activation barriers for structure deformations. Our transition state calculations indicate that the Gibbs free energy barrier is about 2.5 eV for these configurations. The chemical functionalization is not also an appropriate process for gas detection due to poor recovery of the sensor device which is subjected to a strong chemical functionalization. In spite of the all above-mentioned points, we think that the B12N12 can detect the H2CO gaseous molecules through its chemisorption on the exterior surface of the cluster as discussed on here earlier (the configuration B).
It is well known that there is not any universal exchange-correlation functional for all purposes. One of the problems of the functionals is the calculation of HOMO, LUMO and Eg. However, our main quantity is the changes of Eg and not its absolute value. Here, we studied the effect of functionals on the electronic property results. To this aim, we repeated the calculations for the complex B (Fig. 2) with four Minnesota functionals M06-L [37], M06 [38], M06-2X [38], and M06-HF [39] with 0, 27, 54, and 100% Hartree Fock (HF) exchange, respectively. As shown in Table 2, the energy of HOMO, LUMO and Eg is strongly depends on the kind of functional but all functionals show significant change of Eg upon adsorption of formaldehyde which is responsible to the detection process. By increasing the percentage of HF exchange the LUMO and HOMO shift up and down, respectively, thereby increasing the Eg. Also, we have investigated the effect of basis set on the results of ∆G by repeating the calculations for the complex B at X3LYP level with basis sets 6-31+G(d), 6-31++G(d, p), and 6-311++G(d, p). The corresponding calculated ∆G values (Zero point and BSSE corrected) are about -0.54, -0.53, and -0.51 eV, respectively, indicating that enlarging the basis set slightly affects the results.
Table 2 Calculated HOMO, LUMO, and HOMO-LUMO energy gap (Eg) for B12N12 and complex of formaldehyde chemisorption as shown in Fig. 2 by Minnesota functionals (Energies in eV).
| Functional | system | LUMO | HOMO | Eg | ΔEg (%) |
|---|---|---|---|---|---|
| M06-L | B12N12 | -1.25 | -6.21 | 5.82 | 65.41 |
| Complex | -4.20 | -8.18 | 2.01 | ||
| M06 | B12N12 | -0.71 | -7.38 | 7.46 | 44.12 |
| Complex | -3.21 | -9.28 | 4.17 | ||
| M06-2X | B12N12 | 0.17 | -8.48 | 9.45 | 32.08 |
| Complex | -2.06 | -11.24 | 6.41 | ||
| M06-HF | B12N12 | 1.21 | -10.43 | 12.46 | 17.76 |
| Complex | -0.19 | -6.21 | 10.24 |
It should be noted that there is a difference between Eg and band gap. Band gap is defined as the energy difference between the top of the valence band and the bottom of the conduction band in the solid state, but the Eg is defined in molecular level. Matxain et al. [40] have shown that when B12N12 monomers form a solid structure with periodic arrangement, the band gap is somewhat smaller than the Eg of an isolated cluster. Using the generalized gradient approximation (GGA), within the Perdew-Burke-Ernzerhof (PBE) functional, they predicted that the Eg of an isolated B12N12 cluster is about 6.78 eV, while the band gap of the solid state is about 5.20 eV. This indicates that although the band gap of solid state is somewhat smaller than the Eg of the isolated B12N12 monomer, it is still large indicating semiconducting character of the solid structure.
4. Conclusion
We have studied the H2CO adsorption on the exterior surface of B12N12 nanoclusuter, using DFT calculations. It was found that the formaldehyde can be adsorbed on the cluster surface through three different ways including physisorption, chemisorption, and chemical functionalization, with ΔG in the range of -0.07 to -2.00 eV. We showed that the electrical conductivity of the cluster is dramatically changed upon the adsorption of H2CO molecule. Therefore, it is inferred that the B12N12 nanocluster may be a potential gas sensor for detection of H2CO molecule.

