










Servicios Personalizados
Revista
Articulo
 Inglés (pdf)
Inglés (pdf)
 Artículo en XML
Artículo en XML Referencias del artículo
Referencias del artículo
 Enviar artículo por email
Enviar artículo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO -
 Accesos
Accesos
Links relacionados
-
 Similares en
SciELO
Similares en
SciELO
Compartir

 Permalink
PermalinkJournal of the Mexican Chemical Society
versión impresa ISSN 1870-249X
J. Mex. Chem. Soc vol.59 no.4 Ciudad de México oct./dic. 2015
Article
The Role of the π Acceptor Character of Polypyridine Ligands on the Electrochemical Response of Co(II) Complexes and its Effect on the Homogenous Electron Transfer Rate Constant with the Enzyme Glucose Oxidase
Vanessa Ramírez-Delgado,1 Marisela Cruz-Ramirez,1 Luis Felipe Hernández-Ayala,2 Yolanda Reyes-Vidal,1,5 Rita Patakfalvi,3 Juan Carlos García-Ramos,2‡ Francisco J. Tenorio,4 Lena Ruiz-Azuara*2 and Luis Ortiz-Frade1*
1 Departamento de Electroquímica. Centro de Investigación y Desarrollo Tecnológico en Electroquímica S.C. Parque Tecnológico Querétaro, Sanfandila, Pedro de Escobedo, C.P. 76703. Querétaro, México. lortiz@cideteq.mx
2 Laboratorio de Química Inorgánica Medicinal. Departamento de Química Inorgánica y Nuclear, Facultad de Química, Universidad Nacional Autónoma de México, Av. Universidad 3000, Ciudad Universitaria, México, D.F., 04510, México. ‡ Actually as postdoctoral fellow at Departamento de Fisicoquímica, Instituto de Química, UNAM.
3 Universidad de Guadalajara, Centro Universitario de los Lagos. Departamento de Ciencias de la Tierra y de la Vida. Av. Enrique Díaz de León no. 1144, Col. Paseos de la Montaña, Lagos de Moreno, Jalisco, México. CP. 47460.
4 Universidad de Guadalajara, Centro Universitario de los Lagos. Departamento de Ciencias Exactas y Tecnologías. Av. Enrique Díaz de León no. 1144, Col. Paseos de la Montaña, Lagos de Moreno, Jalisco, México. CP 47460.
5 Catedra Conacyt -CIDETEQ.
Received October 7th, 2015;
Accepted February 17th, 2016.
Abstract
In this work the electrochemical behavior of Co(II) complexes with substituted bidentate and tridentate polypyridine ligands [CoL3](BF4)2 and [CoL'2](NO3)2 in 0.1M phosphate buffer pH 7.2 was studied. A reversible electrochemical process Co(II)Ln →Co(III) Ln+1e— was observed. A linear relationship between the redox potential (E°) and the pKa of the non-coordinated ligand was found. It was demonstrated by DFT calculations the use of pKa value as a descriptor of the π acceptor character of a ligand. The electrochemical response in the presence of glucose oxidase (GOx) was also studied. It was possible to establish a tendency between the homogeneous electron transfer rate constant (ks) and the redox potential (E°) for the compounds studied in this work and other examples taken from the literature.
Key words: Co(II) complexes; polypyridine ligands; electrochemistry; redox mediator; glucose oxidase; DFT calculations.
Resumen
En este trabajo se estudió el comportamiento electroquímico de complejos de Co(II) con ligantes polipiridinicos bidentados y tridentados sustituidos [CoL3](BF4)2 y [CoL'2](NO3)2 en soluciones tampón de fosfato pH 7.2 0.1 M. Se observó un proceso electroquímico reversible Co(II)Ln → Co(III)Ln + 1e, encontrándose una relación lineal entre el potencial redox (E°) y el pKa del ligante no coordinado. Mediante cálculos DFT se demostró el uso del valor pKa como descriptor del carácter aceptor π de los ligantes. Se estudió la respuesta electroquímica en presencia de glucosa oxidasa (GOx). Se estableció una tendencia entre la constante de velocidad de transferencia de electrónica homogénea (ks) y el potencial redox (E°) para los compuestos estudiados en este trabajo y otros ejemplos tomados de la literatura.
Palabras clave: Complejos de Co(II); ligantes polipiridínicos; electroquímica; mediadores redox; glucosa oxidasa; cálculos DFT.
Introduction
Octahedral Cobalt complexes with polypyridine ligands have been widely used in several areas of chemistry with technological applications. In the field of dye sensitized solar cells (DSSCs) families of Co(II)/Co(III) complexes with substituted bipyridine, terpyridine and phenathroline ligands have been used as redox mediator for dye regeneration, due to their advantages in comparison with the typical I3/I- electrolyte, such as non-corrosiveness, non-volatility, negligible light absorption, simple electron transfer mechanism and tunable redox potential that increase the maximum open circuit potential (Voc) [1-18].
The non-covalent interactions of the tris-(1,10-phenanthro-line)-Co(III) complex with DNA, demonstrated through cyclic voltammetry, has motived different research groups to develop new electrochemical biosensors based on DNA hybridization. Detection of human immunodeficiency virus (HIV), Escherichia coli, Hepatitis B virus and the chemotherapeutic agent for lymphoblastic leukemia, 6-Mercaptopurine, are some examples of these biosensors [19-35].
In the area of renewable energies using solar radiation, particularly in water splitting to produce H2 and O2, also called artificial photosynthesis, Co(II) complexes with polypyridine ligands have also drawn attention due to the formation of hydride species Co(II)-H and Co(III)-H, required for hydrogen evolution [36-41]. In the realm of molecular electrocatalysis, Co(II)-polypyridine complexes have been proven useful in the electrochemical reduction CO2 and H2O as a potential strategy for energy storage [42-48].
In all these applications a great stability of Co(II) and Co(III) oxidation states either in non-aqueous solvents or in acid and basic media is a necessary condition. Despite the aforementioned versatility and chemical properties of Co(II) polypyridine complexes, these systems have not been used as redox mediators for the development of new reliable and, cheap amperometric glucometers for the control of Diabetes Mellitus. These complexes in comparison with typical redox mediators used in commercial devices, present advantages such as simple methods of preparation and high stability [49]. In the amperometric glucose biosensors the sensibility is controlled by the homogenous electron transfer rate constant (ks) between the oxidized form of the so called "redox mediator" and the reduced form of Glucose oxidase (GOx), where a high rate constant value implies lower amount of enzyme that could lower the cost of the device. Different transition metal complexes, including those with polypyridine ligands have been used as redox mediators [50-61]. It has been suggested that the rate constant (ks) obeys the Marcus cross relation, where one important factor is the difference between the redox potentials of trochemical Response of Co(II) Complexes the couples MIILn/ MIIILn and FAD/FADH2 (-0.22 V vs ENH) for the redox mediator and the active site of the GOx enzyme respectively [55-61]. Nevertheless this topic is still a controversy, because the Marcus cross relation was proposed for metal complexes with outer sphere homogenous electron transfer.
On the other hand, for octahedral low spin Ru(II), Fe(II), Os(II), Mn(II), Cr(II), Rh(II), and Re(II) complexes with polypyridine ligands the redox potential is modulated by the π acceptor character of the ligands [62]. However for Co(II) derivatives, this has not been studied, due the lack of an accurate π acceptor descriptor of high spin complexes. Despite that tunable E° values are desired for specific applications, including their use as redox mediators in glucose biosensors.
Therefore in this work we decided to study the redox behavior of a series of polypyridine Co(II) complexes [CoL3] (BF4)2 and [CoL'2](NO3)2 (L= substituted 1,10-phenanthroline, 2-2'-bipyridine, L'=2,2':6',2"-terpyridine), see scheme 1, in order to establish the role of the n acceptor character of the ligand, using an accurate descriptor, on the modulation of the redox potential (E°) and its effect on the homogenous electron transfer rate constant (ks) between the oxidized form of a redox mediator and the reduced form of GOx. The obtained results are intended to contribute to the understanding of molecular aspects on the design of redox mediators for glucose biosensors based on the modulation of the redox potential for the electron transfer Co(II) Co(II)+ 1e—, that can be extrapolated to other transition metal complexes.
Results and discussion Characterization of complexes
The mixture of the starting salt [Co(H2O)6](BF4)2 or Co(NO3)2 •6H2O with the polypyridine ligand L or L'(see scheme 1) in a molar ratio (1:3) yield yellow powders corresponding to the series of complexes [CoL3](BF4)2 and [CoL'2](NO3)2. The IR spectrum of the [Co(1,10-phenanthroline)3](BF4)2 complex shows typical signals of polypyridine ligand, see fig. 1a. A medium absorption bands at 3068 and 3024 cm-1 attributable to aromatic stretching v (=C-H) are observed. The absorptions bands corresponding to the out of plane C-H bending are registered at 846, 724 cm-1. The stretching bands v (C=C) + v (C=N) occurs at 1604, 1627, 1582 cm-1. A broad signal around 1060 cm-1 characteristic for BF4- anion was also observed. Similar signals are observed in Raman fig. 1b. Additionally vibrational frequencies related to the stretching v(M-N) were recorded at 274 and 226 cm-1, which are in the expected range reported (180-290 cm-1) for Co(II) complexes [63].
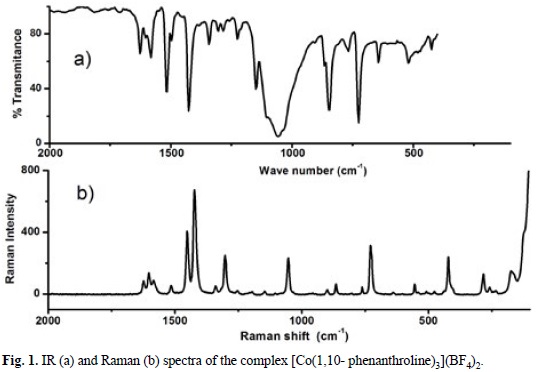
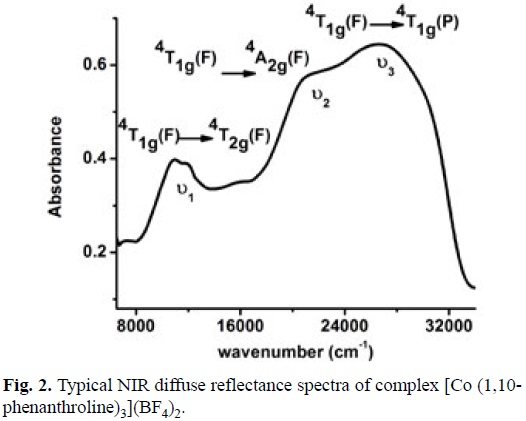
To propose the geometry around the Co(II) center, NIR diffuse reflectance spectra was obtained for this complex, see fig. 2. Three typical electronic transitions for octahedral Co (II) complexes ν1=4T1g (F) →4T2g (F); ν2 = 4T1g (F)→ 4A2g (F); ν3 = 4T1g (F) → 4T1g (P) at 11260, 21770 and 26800 cm-1 respectively were detected [64]. Considering this spectrum and the effective magnetic moment μeff = 4.03 BM (3 unpaired electron), it can be stated unequivocally the formula [Co(1,10-phenanthroline)3](BF4)2. Spectroscopic and magnetic characterization for the other complexes allow us to propose a similar octahedral Co(II) complexes.
Electrochemical behavior of Co(II) complexes with polypyridine ligands in 0.1 M phosphate buffer pH 7.2
In order to explore the electrochemical behavior of the series of Co(II) complexes in physiological conditions, cyclic voltammetry in 0.1 M phosphate buffer pH 7.2 was carried out. Fig. 3a shows a typical voltammogram for [Co(1,10-phenanthroline)3] (BF4)2 obtained with a glassy carbon electrode at 10 mVs-1. One oxidation process (Ia) was detected with a potential peak (Epa) at 0.178 V vs Ag/AgCl. Its corresponding reduction process (Ic) presented a cathodic peak (Epc) at 0.112 V vs Ag/AgCl. A potential peak difference AEp of 0.066 V was calculated. When the scan rate was increased, the anodic peak current (Ia) showed a proportional relation with the v1/2. The behavior mentioned before gives evidence to propose a reversible one electron transfer [Co(II)L3]2+ - [Co(III)L3]3+ + 1e", with a redox potential value E°=0.145 V vs Ag/AgCl [65,66].
One step chronoamperometry experiment stepping the potential from OCP to a value where the process is limited by diffusion was carried out. A typical i(t) vs t-1/2 plot with a linear relationship i(μA)=7.64(μA s1/2) t-1/2 -0.152(μA), r = 0.999, was obtained. From the slope of this equation and according to the Cottrell law, a diffusion coefficient value (DO) of 3.955 x 10-6 cm2 s-1 was calculated [66].
The electrochemical behavior of the other complexes was explored in the same way than that described for the compound [Co(1,10-phenanthroline)3](BF4)2. Similar redox responses were observed with differences in redox potential values being attributed to the electronic properties of the ligands, a summary is presented in table 1. The complex with the highest redox potential corresponds to that where the ligand with the electron-withdrawing substituent (5-nitro-1,10-phenanthroline) is present. On the other hand complexes containing ligands with electron-donating substituents, such as the 5,6-dimethyl-1, 10-phenathroline, decrease the redox potential. In order to understand the electronic properties that have influence over the E° value, we decided to explore a simple and reliable linear relationship.
Linear relationship between the redox potential and the pKa of non-coordinated ligands
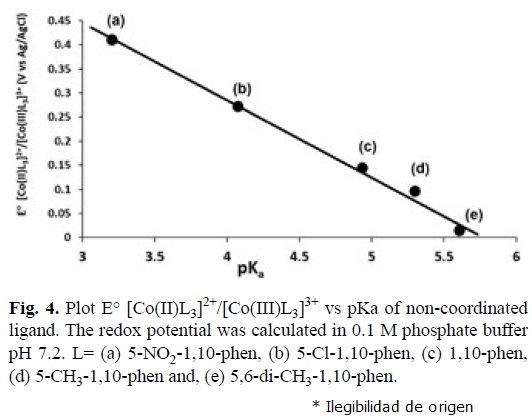
It is reported in the literature, that the pKa of a non-coordinated polypyridine ligand is related with its n acceptor character in metal complexes [67-69]. With this criteria linear relationships between E° and pKa have been described for Cu(II), Fe(II) and Ru(II) complexes, but not for Co(II) compounds. In this work we found this type of relationship for the reversible electron transfer [Co(II)L3]2+ [Co(III)L3]3+ 1e—", see table 1, with the equation E° = -0.1603 pKa + 0.9264 (r=0.999), see fig. 4. Complexes containing ligands with high n acceptor character (low pKa) present high redox potentials. The metal complex with the ligand 2,2'-bipyridine in this relationship gives a low correlation coefficient. This can be a consequence of different electronic and resonant effects due to the presence of only two aromatic rings or low spin/ high spin equilibrium already reported for Cobalt-2,2'-bipyridine complexes [8,10]. Hence we obtained a simple model that predicts the redox behavior of Co(II) complexes, just by knowing the pKa of the free ligand.
Theoretical studies
In order to verify the use of pKa of a non-coordinated ligand as a descriptor of the n acceptor character, theoretical calculations using DFT were carried out. Based on reliable results reported in literature we decided to use the reactivity and response indexes; chemical hardness (η), chemical potential governing charge donating process (μ-), chemical potential governing charge accepting process (μ), electro-donating power (ω-) and electro-accepting power (ω+), which depends on the vertical ionization energy (I) and vertical electron affinity (A), see equations 1-5 [70]



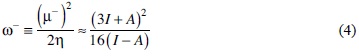
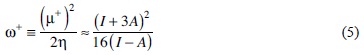
It can be observed that for a low pKa the chemical potential governing charge accepting process (μ+) and electro-accepting power (ω+) presented high values, which confirms the ability of ligands with electron withdrawing substituents to participate in a n back bonding (high capacity to accept charge, due to a high n acceptor character). For the chemical potential governing donation process (μ-) and electro-donating power (ω-), these values are increased when the pKa of the ligand is also increased. This value is related to the o character of the ligand, a necessary condition to participate in the n back bonding [63]. Results are summarized in Table 2.
On the other hand, ligands containing electron donating groups such as methyl or amino group decrease the redox potential of the complex, due to low ionization energy (I). The opposite is observed for ligands with electron withdrawing groups (-Cl and -NO2) where high redox potential values are present (it is more difficult to remove an electron from Co(II) center) as a consequence of the higher ionization energy (I). Fig. 5a, shows a typical HOMO plot of 1,10-phenanthroline ligand, which is very similar for all the calculated derivatives. It can be observed the localization of the molecular orbital over donor atoms. The ligands 2,2'-bypyridine and 2,2':6',2''- ter-pyridine do not present the same tendency, because they have a different electronic delocalization in comparison with 1,10-phenantroline derivatives, see figures 5b and 5c [69].
It was demonstrated that is possible to tune the redox potential with a reliable n acceptor description (pKa of non coordinated ligand). To continue with the aim of this work electrochemical experiments of Co(II) complexes in the presence of GOx are presented in the next section.
Electrochemical behavior of Co(II) complexes with polypyridine ligands in the presence of Glucose Oxidase
Second generation amperometric glucose biosensors, require the presence of a redox mediator, because the active site of Glucose oxidase (FAD) is embedded in the protein domain, and no direct electron transfer between the enzyme and the electrode is recorded [58, 71,72]. The reactions that describe the operation of this type of glucose biosensors are listed below [73]:
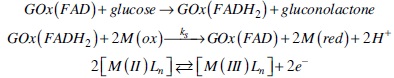
The sensibility of these type of biosensors is evaluated with the homogenous electron transfer rate constant (ks) between the reduced active site of the Glucose oxidase (FADH2) and the oxidized mediator [M(III)Ln]. It has been suggested in literature that this rate constant obeys the Marcus cross relation, see equation 6 and 7, where k11 and k22 values correspond to the self-exchange homogenous electron transfer rate constants for the species involved in the reaction. Whereas the K12 value is the equilibrium constant for the reaction, calculated from the difference of redox potentials between the couples [M(II)Ln] / [M(III)Ln] and FAD/FADH2 (-0.41 V vs Ag/AgCl). The /factor is a parameter that relates k11 and k22 with the collision rate for the reactants, and has a value close to the unity. It should be highlighted that the Marcus cross relation was proposed for outer sphere homogenous electron transfer rate constant for metal complexes. Therefore we decided to study the electrochemical response of three representative cobalt complexes [Co(2,2'-bipyridine)3](BF4)2, [Co(1,10-phenanthroline)3](BF4)2 and [Co(5-chloro-1,10-phenanthroline)3](BF4)2 with different n acceptor ligands in the presence of GOx to evaluate their corresponding ks values and to explore its relationship with the redox potential. Data taken from literature for Os (II) and Ru (II) complexes with the same ligands were also considered in this analysis [50].
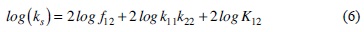

Fig. 6 shows a typical voltammogram of a 1mM solution of [Co(5-Chloro-1,10-phenanthroline)3](BF4)2 in 0.1 M phosphate buffer pH 7.2 with 0.05 M of Glucose in the absence and in the presence of GOx 3.4 |iM at scan rate of 1 mVs-1. When no enzyme was added to the solution a typical Current (I) vs Potential (E) profile for a diffusion control process was recorded, with a maximum current value (Id), fig. 6a. On the other hand in the presence of GOx the voltamogramm presented an S shape catalytic response, with the anodic and cathodic traces superimposed and with a plateau current (Ik) characteristic of ECi' mechanism [74]. To confirm the mechanism and to calculate ks experiments with variable scan rate from 1mV s-1 to 100 mV s-1 were carried out, see fig. 7. The increase in the scan rate makes that the voltammogram pass from an S shape catalytic response (pure kinetic condition) to a partially reversible wave (no substrate consumption) and finally to a reversible wave (no catalysis) [74,75]. According to the method reported by Nicholson and Shain, using a working curve Ψ than depends on the ratio of the catalytic and diffusion currents (Ik/Id) at different scan rates in the presence and absence of GOx respectively. A pseudo-first order catalytic rate constant (kf') was calculated from the slope of Ψ vs v1 plot, see equation 8 [74].


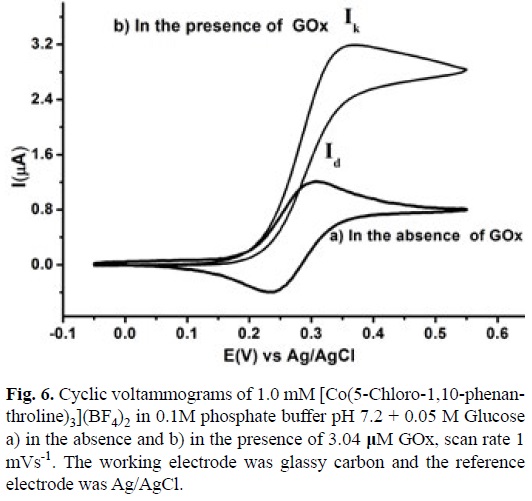
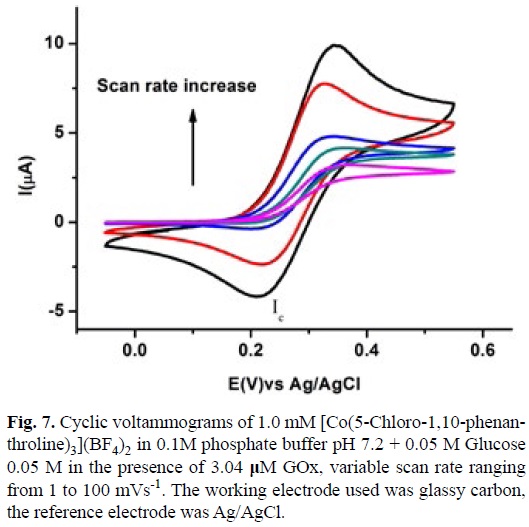
A ks value of 1.75x103 M-1 s-1 was calculated for the homogenous electron transfer between the reduced GOx and [CoIII(5-Cl-1,10-phenanthroline)3]3+, using equation 9. The Enzyme concentration was calculated from value for Ferrocene carboxylic acid, obtained in the same experimental conditions described above and with its corresponding ks value taken form literature [73]. The same experiments were performed for [Co(2,2'-bipyridine)3](BF4)2 and [Co(1,10-phenanthroline)3] (BF4)2, see figures 8 and 9. A summary of this data with the equilibrium constant K12 corresponding to the difference between the redox potential of the mediator and the active site of the enzyme are shown in table 3. Similar analysis with Os (II) and Ru (II) complexes are also present in this table.
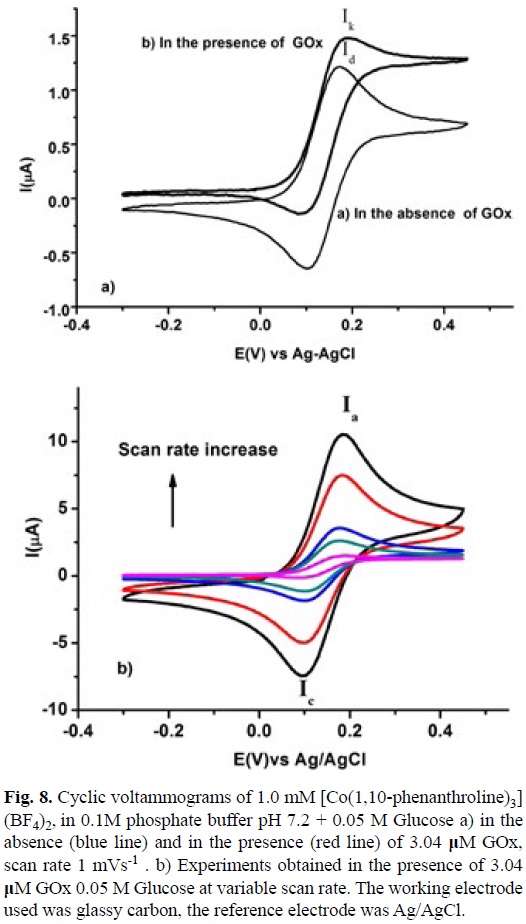
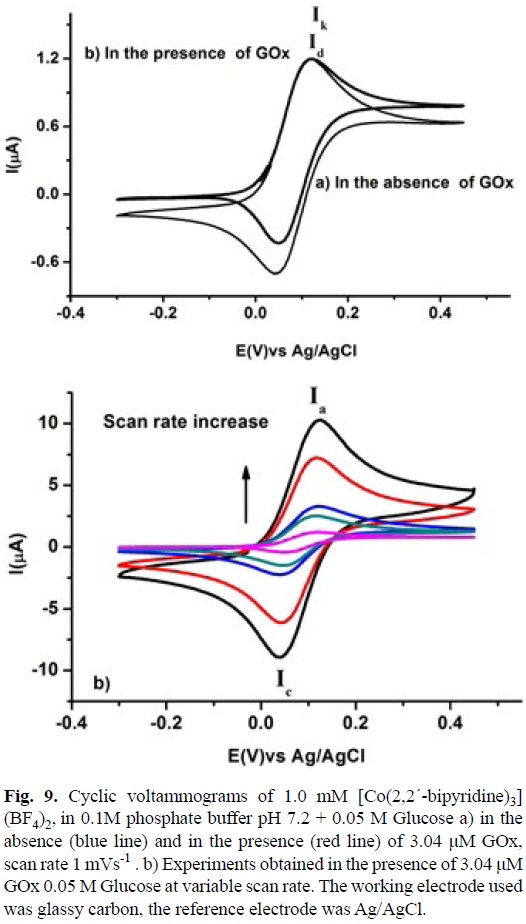
A detailed inspection of table 3, shows that Co(II) complexes, with low redox potential (E°) present low homogenous electron transfer rate constants (ks) and low equilibrium constants K12. High values for both parameters are observed in Ru(II) complexes and intermediate behavior is observed in Os-(II) complexes. Despite no linear relationships between these parameters can be calculated a tendency is observed. The remarkable change in the redox potential values among Co(II), Os(II) and Ru(II) compounds could be explained in terms of simple crystal field theory, keeping in mind the low spin behavior configuration (t2g)5(t2g)2 for Co(II) complexes and the configuration (t2g)6(t2g) for Ru(II) and Os(II) complexes. This consideration implies less energy to oxidize the Co(II) compounds due to lower Crystal Field Stabilization Energy CFSE [63]. Regarding (ks) values, there is not a linear relationship between log ks and log K12. In this case a rough tendency can be observed, which indicates that the Marcus cross relation, equation 6, did not describe correctly this homogenous electron transfer. Actually it is observed that when the log K12 value is increased the parameter log ks presented an asymptotic value around 6.0. This fact suggests that there should be additional factors that control the whole process, such as electrostatic interactions or long distance electronic communication between the active site of the enzyme and the mediator. It should be highlighted that metal complexes with high redox potential (E°) values present high homogenous electron transfer rate constant (ks). Co(II) complexes with a strong π acceptor ligand could be used as a good redox mediator for this kind of amperometric biosensor. These compounds have the advantage of being cheaper compared with their corresponding Os(II) and Ru(II) analogous. The results also show that there is no necessity to use a redox mediator with a high redox potential value due to the limit asymptotic ks value.
Conclusions
A linear relationship between the redox potential E° and the pKa of the non-coordinated ligand for the reversible electron transfer [Co(II)L3]2+ - [Co(III)L3]3++ 1e— with polypyridine ligands was obtained, with the equation E° = -0.1603 pKa + 0.9264 (r = 0.999). It was demonstrated by DFT calculations that the pKa of the non-coordinated ligand is a good descriptor for the π acceptor character of a polypyridine ligand. Co(II) complexes containing ligands with high n acceptor character (low pKa) increase the redox potential E° and the homogenous electron transfer rate constant (ks), which indicates that these compounds could be useful as good redox mediators for glucose biosensors.
Experimental section
Chemicals
All chemicals and solvents in this work were used as received from Aldrich Chemical Co., Acros Organics and J.T. Baker.
Synthesis of the complexes [CoL3BF4].
These metal complexes were synthesized adding dropwise 5 ml of 3 equivalents previously dissolved in methanol to 5 ml 0.02 M of Co(BF4) •6H2O in methanol solution. The reaction mixture was stirred for 2 hours. After this time the solvent was removed on a rotary evaporator at 60°C until a yellow powder was observed. The product was filtered and washed with ethylic ether.
Synthesis of the complex [CoL'2](NO3)2. 1 mmol of Co(NO3)2•6H2O and 3 mmol of the ligand were dissolved separately in methanol. Then the ligand solution was added dropwise to the metallic salt solution. The reaction mixture was stirred for 2 hours. The solvent was removed by slow evaporation until a yellow powder was observed. The product was filtered and washed with ethylic ether.
[Co(1,10-phenanthroline)3](BF4)2. Elemental analysis for CoC36H24N6B2F8, Calc.: %C, 55.9; %H, 3.1; %N, 10.9. ΛM (H2O): 268 Ω-1 cm2 mol-1. μeff 4.07 BM (3 unpaired electron) Found: %C, 55.4; %H, 2.7; %N, 10.9. Mayor IR bands (cm-1): v(=C-H) 3068 3024; v(C=C) + v(C=N) 1627, 1604, 1582; δ(=C-H) 846,724 out of plane; ionic BF4- v(B-F) broad center at 1060. Raman band (cm-1): v(M-N) 274, 226. NIR diffuse reflectance data; 4T1g (F) → 4T2g (F)= 11260, 4T1g (F)→ 4A2g (F) = 21770 and 4T1g (F) → 4T1g (P) = 26800 cm-1
[Co(5-methyl-1,10-phenanthroline)3](BF4)23H2O. Elemental analysis for CoC34H36N6O3B2F8, Calc.: %C, 55.5; %H, 4.9; %N, 10.4. ΛM (H2O): 258 Ω-1 cm2 mol-1. μeff 4.06 BM (4 unpaired electron) Found: %C, 50.9; %H, 4.5; %N, 10.6. Mayor IR bands (cm-1): v(-C-H) 2921, 2867; v(=C-H) 3054, 3096; v(C=C) + v(C=N) 1626, 1602, 1585; δ(=C-H) 897, 876, 804, 813, 734, 724 out of plane; ionic BF4- v(B-F) 1062. Raman band (cm-1): v(M-N) 273, 229. NIR diffuse reflectance data; 4T1g (F) →4T2g (F) = 11260,4T1g (F) → 4T2g (F)= 22060 and 4T1g (F) →4T1g (P) = 27000 cm-1
[Co(4-methyl-1,10-phenanthroline)3](BF4)26H2O. Elemental analysis for CoC39H42N6O6B2F8, Calc.: %C, 50.7; %H, 4.6; %N, 9.1. ΛM (H2O): 268.1 Ω-1 cm2 mol-1. μeff: 4.07 BM (3 unpaired electron) Found: %C, 51.0; %H, 3.8; %N, 9.5. Mayor IR bands (cm-1): v(-C-H) 2918, 2865; v(=C-H) 3065; v(C=C) + v(C=N) 1623, 1604, 1590, 1575; δ(=C-H) 861, 847, 835,786, 727 out of plane; ionic BF4- v(B-F) 1055. Raman band (cm-1): v(M-N) 268, 225. NIR diffuse reflectance data; 4T1g (F) → 4T2g (F) = 11540, % (F) →4A2g (F)= 22900 and 4T1g (F) → 4T1g (P) = 28000 cm-1
[Co(5,6-dimethyl-1,10-1,10-phenanthroline)3](BF4)2 8H2O. Elemental analysis for CoC42H52N6O8B2F8, Calc.: %C, 50.4; %H, 5.2; %N, 8.4. ΛM (H2O): 168.1 Ω-1 cm2 mol-1. μeff: 4.17 BM (3 unpaired electron) Found: %C, 50.7; %H, 4.2; %N, 8.8. Mayor IR bands (cm-1): v(C-H) 2985, 2931, 2874; v(=C-H) 3093; v(C=C) + v(C=N) 1605, 1585; δ(=C-H) 883, 825, 814, 735, 693 out of plane; ionic BF4- v(B-F) 1057. Raman band (cm-1): v(M-N) 281, 236. NIR diffuse reflectance data; 4T1g (F) →4T2g (F) = 11310, 4T1g (F) →4A2g (F)= 22950 and 4T1g (F) → 4T1g (P) = 26800 cm-1
[Co(5-chloro-1,10-phenanthroline)3](BF4)24H2O Elemental analysis for CoC36H29N6O4B2F8 Cl3, Calc.: %C, 45.6; %H, 3.1; %N, 8.9. ΛM (H2O): 168.1 Ω-1 cm2 mol-1. μeff: 4.27 BM (3 unpaired electron) Found: %C, 44.9; %H, 2.7; %N, 9.1. Mayor IR bands (cm-1): v(=C-H) 3043; v(C=C) + v(C=N) 1614, 1604, 1581; δ(=C-H) 891, 876, 825,809, 781, 733, out of plane; ionic BF4- v(B-F) 1060. Raman band (cm-1): v(M-N) 266, 232. NIR diffuse reflectance data; 4T1g (F) → 4T2g (F)= 10290, 4T1g (F) → 4A2g (F)= 22540 and 4T1g (F)→ 4T1g (P) = 27600 cm-1
[Co(5-nitro-1,10-phenanthroline)3](BF4)27H2O. Elemental analysis for CoC30H35N9O12B2F8, Calc.: %C, 41.8; %H, 3.4; %N, 12.2. ΛM (H2O): 168.1 Ω-1 cm2 mol-1. μeff: 4.01 BM (3 unpaired electron) Found: %C, 41.3; %H, 2.7; %N, 12.6. Mayor IR bands (cm-1 ): v(=C-H) 3084; v(C=C) + v(C=N) 1626, 1610, 1587; δ(=C-H) 837, 824, 814, 750, 733, 722 out of plane; v(NO2)sy 1517, v(N=O) 1537; v(NO2)as 1357, 1332; ionic BF4-v(B-F) 1061. Raman band (cm-1): v(M-N) 266, 249. NIR diffuse reflectance data; 4T1g (F) → 4T2g (F)= 10700, 4T1g (F) → 4A2g (F)= 22750 and 4T1g (F) → 4T1g (P) = 27800 cm-1
[Co(5-amine-1,10-phenanthroline)3](BF4)23H2O Elemental analysis for CoC36H33N9O3B2F8, Calc.: %C, 49.6; %H, 3.8; %N, 14.4. ΛM (H2O): 168.1 Ω-1 cm2 mol-1. μeff: 4.2 BM (3 unpaired electron) Found: %C, 50.1; %H, 2.9; %N, 14.1. Mayor IR bands (cm-1): v(C-H) 3048; v(N-H) 3442, 3390; v(C=C) + v(C=N) 1640, 1616, 1596, 1517; δ(=C-H) 853, 823, 803, 731, 723 out of plane; ionic BF4- v(B-F) 1056. Raman band (cm-1): v(M-N) 274 and 226.NIR diffuse reflectance data; 4T1g (F) → 4T2g (F)= 11000, 4T1g (F) →4A2g (F)= 21000 and 4T1g (F)→4T1g (P) = 26000 cm-1
[Co(2,2'-bipyridine)3](BF4)2.7H2O Elemental analysis for CoC30H36N6 O6B2F8, Calc.: %C, 44.5; %H, 4.5; %N, 10.4. ΛM (H2O): 168.1 Ω-1 cm2 mol-1. μeff: 4.03 BM (3 unpaired electron) Found: %C, 44.4; %H, 4.0; %N, 10.5. Mayor IR bands (cm-1): v(=C-H) 3098, 3072, 3053, 3027; v(C=C) + v(C=N) 1604, 1596, 1574, 1566; δ(=C-H) 774, 736 out of plane; ionic BF4- v(B-F) 1058. Raman band (cm-1): v(M-N) 236. NIR diffuse reflectance data; 4T1g (F) → 4T2g (F)= 11780, and 4T1g (F)→4T1g (P) = 24600 cm-1
[Co(2,2':6',2''-terpyridine)2](NO3)2 4H2O Elemental analysis for CoC30H30N8O10 Calc.: %C, 49.9; %H, 4.2; %N, 15.5. ΛM (H2O): 168.1 Ω-1 cm2 mol-1. μeff: 4.01 BM (1 unpaired electron) Found: %C, 50.1; %H, 4.0; %N, 15.1. Mayor IR bands (cm-1 ): v(C-H) 3076, 3062, 3040; v(C=C) + v(C=N) 1600, 1574, 1159; δ(=C-H) 829, 773, 732 out of plane; ionic NO3- 1333. Raman band (cm-1): v(M-N) 240.
Theoretical calculations
Density functional theory [76-78] as implemented in Gaussian 09 [79] was used for all calculations for single ligand molecules. Full geometry optimization without symmetry constraints was performed using the three-parameter B3LYP [80-82] density functional and the 6-311+G (d,p) [83] basis set. Optimized geometries of local minima were verified by the number of imaginary frequencies (which should be zero). Previous studies indicate that DFT results are very good describing stabilities and equilibrium geometries when using hybrid functionals which include partially the Hartree-Fock exchange energy and are consistent with those obtained using the Mõller-Plesset perturbational theory at second order and basis sets of medium quality, such as 6-31G(d,p), and cc-pVDZ [84].
Physical measurements
Elemental analyses were carried out using a Fissons Instruments Analyzer model EA 1108 using a sulfanilamide standard. IR spectra were acquired with a Nexus Thermo Nicolet spectrophotometer in the spectral range 4000 a 400 cm-1 on KBr disks. Raman spectroscopy measurements were obtained with a Thermo Scientific DXR Raman microscope, with a 780 nm laser (4 mW power out) in the range from 100 to 3500 cm-1. Electronic spectra were recorded on Thermo Evolution Array spectrophotometer (200-1100 nm). Solid state UV-Vis-NIR spectra were recorded (40000-4000 cm-1) on a Cary-5E (Varian) spectrophotometer. Conductivity measurements were performed using an YSI conductimeter model 302, with parallel plates (Φ=1cm-1). Magnetic susceptibility measurements were recorded at room temperature on a Johnson-Matthey DG8 5HJ balance, using the Gouy method.
Electrochemical studies
Electrochemical measurements were carried out with a Potentiostat/Galvanostat Biologic SP-50, at sample concentrations of 1x10-3 M in 0.1 M phosphate buffer pH 7.2. A typical three electrode array was employed. A glassy carbon disk (Φ = 3 mm) was used as working electrode. A Platinum served as counter-electrode and, a commercial Ag/AgCl electrode was used as reference. Prior each experiment the working electrode was polished with a Alumina (0.3 μm), rinsed and placed on an ultrasonic bath. The solutions were also bubbled with nitrogen for 10 minutes before starting the electrochemical measurements. Cyclic voltammetry was initiated from open circuit potential in positive and negative direction using a scan rate from 10 to 1000 mVs-1. One step chronoamperometry experiments were obtained by stepping the potential from open circuit potential (E1) to value E2 where the electrochemical process is limited by diffusion, with a perturbation time of 1s. Current interrupt method was used for ohmic drop (IR) compensation.
For the homogenous electron transfer rate constant (redox mediation) experiments the sample concentration of metal complexes was 1x10-3 M in 0.1M phosphate buffer pH 7.2+ 0.05 M of Glucose (prepared from a 1M stock solution stored overnight) and 3.04 μM of Glucose Oxidase (EC 1.1.3.4 type II, from Aspergillus Niger molecular weight 186000 Oriental Yeast Co., LTD). Enzyme concentration was calculated from homogenous electron transfer rate constant ks = 2.01x105 (M-1 s-1) experiments using Ferrocene carboxylic acid as reference, according to the literature [73]. This was also confirmed with a spectrophotometric method reported before [85].
Acknowledgement
The authors thank CONACyT (130500), UNAM-PAPIIT (217613) and UNAM-PAIP (3590-19) for financial support; VRD, and JCGR thank CONACYT and FARMED/CONACYT for the scholarships.
References
1. Lee, D. K.; Ahn K-S.; Thogiti, S.; Kim, J. H., Dyes and Pig. 2015, 117, 83-91. [ Links ]
2. Sun, Z-Z.; Li, Q-S.; Zhang, M.; Li, Z-S., J. Power Sources 2015, 294, 264-271. [ Links ]
3. Kirner, J.T.; Elliott, C.M., J. Phys. Chem., 2015, 119, 17502-17514. [ Links ]
4. Shen, C.; Wang, X.; Jiang, X-F.; Zhu, H.; Li, F.; Yang, J.; Xu, Q-H.; Wang, Q., J. Phys. Chem., 2015, 119, 9774-9781. [ Links ]
5. Jung, H.; Koo, B.; Kim, J-Y.; Kim, T.; Son, H.J.; Kim, B-S.; Kim, J.Y.; Lee, D-K.; Kim, H.; Cho, J.; Ko, M.J., ACSAppl. Mater. Interfaces 2014, 6, 19191-19200. [ Links ]
6. Ondersma, J. W. ; Hamann, T. W., Coord. Chem. Rev. 2013, 257, 1533-1543. [ Links ]
7. Nolana, J. P.; Jones, T. W.; Donne, S. W.; Wilsona, G. J., Electrochim. Acta, 2013, 108, 690-697. [ Links ]
8. Salvatori, P.; Marotta, G.; Cinti, A.; Mosconi, E.; Panigrahi, M.; Giribabu, L.; Nazeeruddin, M.K.; De Angelis, F., Inorg. Chim. Acta, 2013, 13, 106-112. [ Links ]
9. Mba, M.; D'Acunzo, M.; Salice, P.; Carofiglio, T.; Maggini, M.; Caramori, S.; Campana, A.; Aliprandi, A.; Argazzi, R.; Carli, S.; Bignozzi, C.A., J. Phys. Chem., 2013, 117, 19885-19896. [ Links ]
10. Mosconi, E.; Yum, J-H.; Kessler, F.; Gómez García, C.J.; Zuccaccia, C.; Cinti A.; Nazeeruddin, M.K.; Gratzel, M.; De Angelis, F., J. Am. Chem. Soc., 2012, 134, 19438-19453. [ Links ]
11. Zong X.; Liang M.; Fan C.; Tang K.; Li G.; Sun, Z.; Xue, S., J. Phys. Chem., 2012, 116, 11241-11250. [ Links ]
12. Feldt, S.M.; Wang, G.; Boschloo, G.; Hagfeldt, A., J. Phys. Chem., 2011, 115, 21500-21507. [ Links ]
13. Bai, Y.; Zhang, J.; Zhou, D.; Wang, Y.; Zhang, M.; Wang, P., J.Am. Chem. Soc., 2011, 133, 11442-11445. [ Links ]
14. Kavan, L.; Yum, J-H.; Gratzel, M., Nano Lett., 2011, 11, 5501-5506. [ Links ]
15. Kavan, L.; Yum, J-H.; Nazeeruddin, M.K.; Gratzel M., ACS Nano, 2011, 11, 9171-9178. [ Links ]
16. DeVries, M.J.; Pellin, M.J.; Hupp, J.T., Langmuir, 2010, 26, 11, 9082-9087. [ Links ]
17. Lee, H.J.; Chen, P.; Moon, S-J.; Sauvage, F.; Sivula, K.; Bessho, T.; Gamelin, D.R.; Comte, P.; Zakeeruddin, S.M.; Seok, S.; Gratzel, M.; Nazeeruddin, Md. K., Langmuir, 2009, 25, 7602-7608. [ Links ]
18. Sapp, S.A.; Elliott, C.M.; Contado, C.; Caramori, S.; Bignozzi, C.A., J. Am. Chem. Soc., 2002, 124, 11215-11222. [ Links ]
19. Carter, M.T.; Rodriguez, M.; Bard A.J., J. Am. Chem. Soc, 1989, 111, 8901-8911. [ Links ]
20. Niu, S.; Sun, J.; Nan, C.; Lin, J., Sens. ActuatorsB, 2013, 176, 58-63. [ Links ]
21. Sheng-Zhen, C.; Qiang, C.; Fang-Yi, P.; Xin-Xin, H.; Yu-Ling, J., Chin. J. Anal. Chem., 2012, 40, 1194-1200. [ Links ]
22. Santiago-Berríos, M.B.; Declet-Flores, C.; David, A., Borrero, S.; Vélez, M.M.; Díaz-Díaz, A.; Guadalupe, A.R.; Colón, J.L., Langmuir, 2012, 4447-4452. [ Links ]
23. Liu, A.; Wang K.; Weng, S.; Lei, Y.; Lin, L.; Chen, W.; Lin, X.; Chen, Y., Trend. Anal. Chem., 2012, 37, 101-111. [ Links ]
24. Zhou, P.; He, L.; Gan, G.; Ni, S.; Li, H.; Li, W., J. Electroanal. Chem., 2012, 665, 63-69. [ Links ]
25. Xu, S.; Lu C.; Shao, J.; Li, Q.; Li, H.; Li, W., J. Electroanal. Chem., 2011, 661, 287-293. [ Links ]
26. Liu, X.; Qu, X.; Fan, H.; Ai, S.; Han, R., Electrochim. Acta, 2010, 55, 6491-6495. [ Links ]
27. Li, F.; Chen, W.; Tang, C.; Zhang, S., Talanta, 2008, 77, 1-8. [ Links ]
28. Niu, S-Y.; Zhang, S-S.; Wang, L.; Li, X.M., J. Electroanal. Chem., 2006, 597, 111-118. [ Links ]
29. Erdem, A.; Ariksoysal, D.O.; Karadeniz, H.; Kara, P.; Sengonul, A.; Sayiner, A.A.; Ozsoz, M., Electrochem. Comm., 2005, 7, 815-820. [ Links ]
30. Elicia, L.S. Wong Mearns, F.J.; Gooding, J.J., Sens. Actuator., 2005, 111-112, 515-521. [ Links ]
31. Jin, B.; Ji, X.; Nakmura, T., Electrochim. Acta, 2004, 50, 1049-1055. [ Links ]
32. Zhang, R-Y.; Pang, D-W.; Zhang, Z-L.; Yan, J-W.; Yao, J-L.; Tian, Z-Q.; Mao B-W.; Sun, S-G., J. Phys. Chem, 2002, 106, 11233-11239. [ Links ]
33. Ji, L-N.; Zou, X-H.; Liu, J-G., Coord. Chem. Rev., 2001, 216-217, 513-536. [ Links ]
34. a)Iuni-Iui, Z.; Hong C.; Ruifu, Y., Biotechnol Adv., 1997, 15, 4358. [ Links ] b) Wang, J.; Cai, X.; Rivas, G.; Shiraishi, H.; Farias, P.A.M.; Dontha, N., Anal. Chem., 1996, 68, 2629-2634. [ Links ]
35. Wang, J.; Palecek, E.; Nielsen, P.E.; Rivas, G.; Cai, X.; Shiraishi, H.; Dontha, N.; Luo, D.; Farias, P.A.M., J. Am. Chem. Soc., 1996, 118, 7667-7670. [ Links ]
36. Tong, L.; Kopecky A.; Zong, R.; Gagnon, K.J.; Ahlquist, M.S.G.; Thummel, R.P., Inorg. Chem., 2015, 54, 7873-7884. [ Links ]
37. Sasaki, Y.; Kato, H.; Kudo, A., J. Am. Chem. Soc., 2013, 135, 5441-5449. [ Links ]
38. Queyriaux, N.; Jane, R.T.; Massin, J.; Artero, V.; Chavarot-Kerlidou, M.; Queyriaux, N., Coord. Chem. Rev, 2015, 46, 1-17. [ Links ]
39. Eckenhoff, W.T.; McNamara, W.R.; Du, P.; Eisemberg, R., Biochim. Biophys. Acta, 2013, 1827, 958-973. [ Links ]
40. Losse, S.; Vos J.G.; Rau, S., Coord. Chem. Rev, 2010, 254, 2492-2504. [ Links ]
41. Chen, X.; Ren, H.; Peng, W.; Zhang, H.; Lu, J.; Zhuang, L., J. Phys. Chem., 2014, 118, 20791-20798. [ Links ]
42. Zhang, P.; Wang, M.; Gloaguen, F.; Chen, L.; Quentel, F.; Sun, L., Chem. Commun., 2013, 49, 9455. [ Links ]
43. Ward, A.L. ; Elbaz, L. ; Kerr, J.B.; Arnold, J., Inorg. Chem., 2012, 51, 4694-4706. [ Links ]
44. Isaacs, M.; Canales, J.C.; Aguirre, M.J.; Estiu, G.; Caruso, F.; Ferraudi, G.; Costamagna, J., Inorg. Chim. Acta, 2002, 339, 224-232. [ Links ]
45. Chiaricato, G. Jr.; Arana, C.R.; Casado, C.; Cuadrado, I.; Abruña, H.D., Inorg. Chim. Acta, 2000, 300-302, 32-42. [ Links ]
46. La, K-M.; Wong, K-Y.; Yang, S-M. ; Che, C-M., J. Chem. Soc. Dalton Trans, 1995, 1103-1107. [ Links ]
47. Arana, C.; Yan, S.; Keshavarz, K. M.; Potts, K.T.; Abruña, H.D., Inorg. Chem., 1992, 31, 3680-3682. [ Links ]
48. Simpson, T.C.; Durand, R.R. Jr., Electrochim. Acta, 1988, 33, 581-583. [ Links ]
49. Heller, A.; Feldman, B., Chem. Rev., 2008, 108, 2482-2505. [ Links ]
50. Zhang, C.X.; Haruyama, T.; Kobatake, E.; Aizawab, M., Anal. Chim. Acta, 2000, 408, 225-232. [ Links ]
51. Harwood, G.W.J.; Pouton, C.W., Adv. Drug Deliver Rev., 1991, 18, 163-191. [ Links ]
52. Nakabayashi, Y.; Nakamura, K.; Kawachi, M.; Motoyama, T.; Yamauchi O., J. Biol. Inorg. Chem., 2003, 45-52. [ Links ]
53. Nakabayashi, Y.; Hirosaki, Y.; Yamauchi, O., Bioelectrochem., 2006, 69, 216-222. [ Links ]
54. Shklover, V.; Nesper, R.; Zakeeruddin, S.M.; Fraser, D.M.; Gratzel, M., Inorg. Chim. Acta, 1996, 247, 237-245. [ Links ]
55. Castillo, J.; Gáspár, J.; Leth, S.; Niculescu, M.; Mortari, A.; Bon-tidean, I.; Soukharev, V.; Dorneanu, S. A.; Ryabov, A. D.; Csõregi, E., Sens. Actuactor. B, 2004, 102, 179-194. [ Links ]
56. Johnson, J.M; Halsall, H.B.; Heineman ,W.R., Anal. Biochem, 1983, 133, 186- 189. [ Links ]
57. DiVirgilio-Thomas, J.M.; Heineman, W.R.; Seliskar, C.J., Anal. Chem., 2000, 72, 3461-3467. [ Links ]
58. Vashist, S.; Zhenga, K.; Al-Rubeaan, D.; Luong, K.; Sheu, J.H., Anal. Chim. Acta, 2011, 703, 124-136. [ Links ]
59. Zakeeruddin, S.M.; Fraser, D.M.; Nazeeruddin, M-K.; Gratzel, M., J. Electroanal. Chem, 1992, 337, 253-283. [ Links ]
60. Kurova, V. S.; Ershov, A. Yu.; Ryabov, A. D., Russ. Chem. Bull., Int. Ed, 2001, 10, 1849-1854. [ Links ]
61. Chaubey, A.; Malhotra, B.D., Biosens. Bioelectron., 2002, 17, 441-456. [ Links ]
62. Lever, A. B. P., Inorg. Chem. 1990, 29, 1271-1285. [ Links ]
63. Nakamoto, K., Infrared spectra of inorganic and coordination compounds. 2da ed.; Wiley: 1963. [ Links ]
64. Huheey, J.E.; Keiter, E.A. ; Keiter, R.L. Inorganic Chemistry: Principles of Structure and Reactivity, Harper Collins College Publisher, New York, 1993. [ Links ]
65. Zanello, P.; Chemistry, R. S. o., Inorganic Electrochemistry: Theory, Practice and Applications. Royal Society of Chemistry: Universidad de Siena, Italia, 2003. [ Links ]
66. Bard, A. J.; Faulkner, L. R. Electrochemical Methods, Fundamentals and Applications; Ed. John Wiley and Sons, 1980. [ Links ]
67. Gasque, L.; Medina, G.; Ruiz-Ramirez, L.; Moreno-Esparza, R., Inorg. Chim. Acta 1999, 288, 106-111. [ Links ]
68. Ortiz-Frade, L.A.; Ruiz-Ramírez, L.; González, I.; Marín-Becerra, A.; Alcarazo, M.; Alvarado-Rodriguez, J.G.; Moreno-Esparza R., Inorg. Chem. 2003, 42, 1825-1834. [ Links ]
69. García-Ramos, J.C.; Galindo-Murillo, R.; Tovar-Tovar, A.; Alonso-Saenz, A.L.; Gómez-Vidales, V.; Flores-Álamo, M.; Ortiz-Frade, L.; Cortes-Guzmán, F.; Moreno Esparza, R.; Campero A.; Ruiz-Azuara, L. Chem. Eur. J. , 2014, 20, 13730-13741. [ Links ]
70. a) Gazquez, J. L.; Cedillo, A.; Vela, A., J. Phys. Chem. A., 2007, 111, 1966-1970 b) Gazquez, [ Links ] J. L., J. Mex. Chem. Soc., 2008, 52, 3-10 c) Ramí [ Links ]rez-Ramírez, J. Z.; Vargas, R.; Garza, J.; Gazquez, J. L., J. Phys. Chem. A., 2010, 114,7945-7951. [ Links ]
71. Wang, J., Electroanal. 2001, 13, 983-988. [ Links ]
72. Newman, J. D.; Turner, A. P. F., Biosens. Bioelectron. 2005, 20, 2435-2453. [ Links ]
73. Cass, A. E.; Davis, G.; Francis, G. D.; Hill, H. A. O.; Aston, W. J.; Higgins, I. J.; Plotkin, E. V.; Scott, L. D.; Turner, A. P., Anal. Chem. 1984, 56, 667-671. [ Links ]
74. Nicholson, R. S.; Shain, I.,Anal. Chem, 1964, 36, 706-723. [ Links ]
75. Savéant, J. M., Elements of Molecular and Biomolecular Electrochemistry: An Electrochemical Approach to Electron Transfer Chemistry. Wiley: 2006. [ Links ]
76. Kohn, W.; Becke, A. D.; Parr, R. G.; J. Phys. Chem., 1996, 100, 12974-12980. [ Links ]
77. Hohenberg, P.; Kohn, W.; Phys. Rev, 1964, 136, B864-B871. [ Links ]
78. Kohn, W.; Sham, L.; J. Phys. Rev, 1965, 140, A1133-A1138. [ Links ]
79. Gaussian 09, Revision D.01, Frisch, M. J.; Trucks, G. W.; Schlegel, H. B.; Scuseria, G. E.; Robb, M. A.; Cheeseman, J. R.; Scalmani, G.; Barone, V.; Mennucci, B.; Petersson, G. A.; Nakatsuji, H.; Caricato, M.; Li, X.; Hratchian, H. P.; Izmaylov, A. F.; Bloino, J.; Zheng, G.; Sonnenberg, J. L.; Hada, M.; Ehara, M.; Toyota, K.; Fukuda, R.; Hasegawa, J.; Ishida, M.; Nakajima, T.; Honda, Y.; Kitao, O.; Nakai, H.; Vreven, T.; Montgomery, J. A., Jr.; Peralta, J. E.; Ogliaro, F.; Bearpark, M.; Heyd, J. J.; Brothers, E.; Kudin, K. N.; Staroverov, V. N.; Kobayashi, R.; Normand, J.; Raghavachari, K.; Rendell, A.; Burant, J. C.; Iyengar, S. S.; Tomasi, J.; Cossi, M.; Rega, N.; Millam, J. M.; Klene, M.; Knox, J. E.; Cross, J. B.; Bakken, V.; Adamo, C.; Jaramillo, J.; Gomperts, R.; Stratmann, R. E.; Yazyev, O.; Austin, A. J.; Cammi, R.; Pomelli, C.; Ochterski, J. W.; Martin, R. L.; Morokuma, K.; Zakrzewski, V. G.; Voth, G. A.; Salvador, P.; Dannenberg, J. J.; Dapprich, S.; Daniels, A. D.; Farkas, Õ.; Foresman, J. B.; Ortiz, J. V.; Cioslowski, J.; Fox, D. J. Gaussian, Inc., Wallingford CT, 2009. [ Links ]
80. Becke, A. D.; Phys. Rev. A, 1988, 38, 3098-3100. [ Links ]
81. Mielich, B.; Savin, A.; Stoll, H.; Peuss, H.; Chem. Phys. Lett., 1989, 157, 200-206. [ Links ]
82. Lee, C.; Yang, W.; Parr, R. G.; Phys. Rev. B., 1988, 37, 785-789. [ Links ]
83. (a) Krishnan; R.; Binkley; J. S.; Seeger R.; Pople, J. A. J. Chem. Phys, 1980, 72, 650. [ Links ] (b) Clark T.; Chandrasekhar, J.; Spitznagel, G. W.; Schleyer, P. v. R. J. Comput. Chem., 1983, 4, 294. [ Links ] (c) Frisch, M. J.; Pople, J. A.; Binkley, J. S. J. Chem. Phys, 1984, 80, 3265. [ Links ] (d) McLean, A. D.; Chandler, G. S. J. Chem. Phys. 1980, 72, 5639.
84. Shishkin, O. V.; Gorb, L.; Luzanov, A. V.; Elstner, M.; Suhai, S.; Leszczynski, J.; J. Mol. Struct. (Theochem), 2003, 625, 295-303. [ Links ]
85. Duke, R.F; Weibel, M.K.; Page, D.S,; Bulfrin, V.G.; Luthy, J. J. Am. Chem. Soc., 1969, 91, 3904-3909. [ Links ]

