










Servicios Personalizados
Revista
Articulo
 Inglés (pdf)
Inglés (pdf)
 Artículo en XML
Artículo en XML Referencias del artículo
Referencias del artículo
 Enviar artículo por email
Enviar artículo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO -
 Accesos
Accesos
Links relacionados
-
 Similares en
SciELO
Similares en
SciELO
Compartir

 Permalink
PermalinkJournal of the Mexican Chemical Society
versión impresa ISSN 1870-249X
J. Mex. Chem. Soc vol.59 no.2 Ciudad de México abr./jun. 2015
Articles
Synthesis of Fluorescent oligo(p-phenyleneethynylene) (OPE3) via Sonogashira Reactions
Mariana Flores-Jarillo,1 Francisco Ayala-Mata,2 Gerardo Zepeda-Vallejo,2 Rosa Ángeles Vázquez-García,3 Gabriel Ramos-Ortiz,4 Miguel Ángel Méndez-Rojas,5 Oscar Rodolfo Suárez-Castillo,1 and Alejandro Alvarez-Hernández1*
1 Área Académica de Química, Universidad Autónoma del Estado de Hidalgo, Carr. Pachuca Tulancingo Km 4.5, Pachuca, Hidalgo 42184, México. * Corresponding author: Alejandro Alvarez-Hernández, email: alvarez@uaeh.edu.mx, alalvareher@yahoo.com
2 Departamento de Química Orgánica, Escuela Nacional de Ciencias Biológicas, Instituto Politécnico Nacional, Prol. Carpio y Plan de Ayala s/n, 11340 México, D.F., México.
3 Área Académica de Ciencias de la Tierra y Materiales, Universidad Autónoma del Estado de Hidalgo, Carr. Pachuca Tulancingo km 4.5, Pachuca, Hidalgo 42184, México.
4 Centro de Investigaciones en Óptica. A.P. 1-948, León, México.
5 Departamento de Química y Biología, Universidad de las Américas-Puebla, Ex-Hda de Sta. Catarina Mártir, A. P. 100, Cholula 72820, Puebla, México.
Received February 16th, 2015
Accepted May 28th, 2015
Abstract
Sonogashira reactions of 4-(2,5-diiodobenzoyl)morpholine and 4-(5-bromo-2-iodobenzoyl)morpholine with arylacetylenes catalyzed by Pd2(dba)3 in DMSO allowed preparation of fluorescent oligo(p-phenyleneethynylene)s (OPE3) with fluorescence quantum yields up to 0.87. DMSO proved to be very efficient for this double Sonogashira coupling in which other solvents failed.
Key words: Sonogashira coupling, oligo(p-phenyleneethynylene)s, OPE3, fluorescence.
Resumen
Las reacciones de Sonogashira de la 4-(2,5-diiodobenzoil)morfolina y la 4-(5-bromo-2-iodobenzoil)morfolina con arilacetilenos catalizadas por Pd2(dba)3 en DMSO permitieron preparar oligo(p-fenilenetinilenos) (OPE3) fluorescentes con rendimientos cuánticos de hasta 0.87. El DMSO demostró ser muy eficiente para este doble acoplamiento de Sonogashira en el cual otros disolventes no fueron adecuados.
Palabras clave: Acoplamiento de Sonogashira, oligo(p-fenilenetinilenos), (OPE3), fluorescencia.
Introduction
The Sonogashira [1] coupling reaction and its variants [2] have found widespread use in the synthesis of oligomeric arylenethynylene materials [3] These conjugated materials incorporate aryl, heteroaryl [4] and organometallic [5] units joined by acetylene links and display remarkable electronic and optical properties [6], such as semiconduction and fluorescence, for which they have been used to construct molecular wires [7], chemosensors [8], imaging materials [9] and the emitting layer in light emitting devices [10]. Short oligomers [11] and small molecules [12] of precise structure can retain some of the electronic and optical properties of their polymeric counterparts and are more easily manipulated and prepared. Thus, in this work a series of oligo(p-phenyleneethynylene)s OPE3s (Fig. 1) was prepared in search for highly fluorescent molecules of low molecular weight for future adaptation and application as biological probes. Herewith the synthesis of these compounds by double Sonogashira couplings in DMSO and their optical properties are described.

At the onset of this work, OPE3 compounds of type 1 were designed as low molecular weight models of fluorescent polyphenylenethynylenes [13]. Substituents on the terminal aryl groups allow tuning of the optical properties and the morpholine amide group at the central aryl unit was designed to confer solubility to phenyleneethynylenes in organic solvents. The synthesis of pseudo symmetrical compounds was envisaged as a double Sonogashira coupling of diiodoamide 2 with two equivalents of an arylacetylene 3. (Scheme 1)

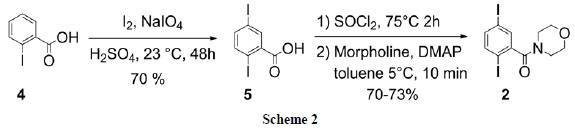
Thus, synthesis of the required diiodoarene 2 started with iodination of commercial 2-iodobenzoic acid 4 with iodine/NaIO4 [14] in sulfuric acid that led to diiodo acid 5, which was converted to 2 by reaction with thionyl chloride followed by addition of morpholine and catalytic DMAP (Scheme 2).
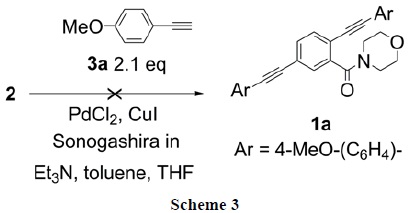
Coupling of diiodoarene 2 with two equivalents of 4-ethynylanisole (3a) to produce 1a was chosen as model reaction to establish a protocol for preparation of a set of related OPE3s 1. Unexpectedly, the reaction proved to be problematic under standard Sonogashira conditions (PdCl2, CuI and triphenylphosphine) in toluene, THF and triethylamine (Scheme 3).
Thus, it was decided to study the possible effect of other solvents in this Sonogashira coupling. Thus, coupling of 2 and 3a was carried out using several organic solvents of different polarity [15] (Scheme 4) and Pd2(dba)3 [16] was used as the source of catalyst. Results are summarized in Table 1. Reaction in Et3N (entry 1) gave a mixture of OPE3 1a and monoalkyne 6a, whose structure was elucidated by HMBC and X-ray diffraction studies. Reactions in toluene (entry 2) and THF (entry 3) were sluggish. Coupling in anhydrous DMF (entry 4) gave products 1a (double coupling) and 6a (monocoupling) in almost equal yields. Remarkably, reaction in anhydrous DMSO (entry 5) proceeded in 1 h and gave exclusively the desired double-coupled product 1a in good yield. Couplings in acetonitrile (entry 6) and nitromethane (entry 7) yielded mixtures of mono (minor) and double coupling (major) products. Ulven and coworkers [17] have reported the beneficial effect of addition of water to TMEDA used as solvent in a case of a problematic Sonogashira coupling. Accordingly, the use of a 9:1 TMEDA-water mixture was tested (entry 8). However, this reaction gave a mixture of coupling products 1a, 6a and 7a in low yield. Thus, of the solvents studied only DMSO was effective for the model double Sonogashira coupling.
It was explored if adventitious water in DMSO represented any problem in these Sonogashira couplings since this solvent is hygroscopic and also known for remarkable changes of its properties in the presence of water [18]. Consequently, model experiments in DMSO containing 5, 33 and 67% of water were carried out (Scheme 5) and their results are shown in Table 2. While coupling of 2 and 3a in commercial “anhydrous DMSO" (0.013% H2O) gave 81% of 1a (Table 2, entry 1) the same reaction in DMSO containing 5% water led to a 10% improved yield (entry 2; both coupling reactions in dry and wet DMSO were repeated at least three times). The reaction in DMSO containing 33% water led to a significant decrease in yield of 1a and formation of monocoupled 6a as the major product (entry 3). Reaction in 33% DMSO - 67% H2O (entry 4) led to reactant solubility problems and gave almost equal amounts of mono (6a) and double (1a) coupled products. In addition, Sonogashira couplings with other arylacetylenes were tested in 5:95 water-DMSO (entries 5-10). The results show that couplings using arylacetylenes substituted with electron donating groups OCH3 (entry 1 vs. entry 2) and Me2N (entry 5 vs. entry 6) improved their chemical yield by about 10% in 5% water-DMSO vs. reaction in dry DMSO. However, water had little effect in chemical yield of coupling reactions with arylacetylenes substituted with electron withdrawing groups such as CN (entries 7 and 8), NO2 (entries 9 and 10) and methylketone (entries 11 and 12).
The observed solvent effect in these coupling reactions could be due to coordination of DMSO with Pd. The coordinating ability of DMSO towards Pd is known [19] and it has been associated to keep catalytically active Pd° in solution, thus avoiding formation of black palladium which is inactive as catalyst. Other examples of Pd catalyzed alkynylation reactions in DMSO are known [20] Studies of some other reactions involving the use of Pd and DMSO show the active role of this solvent in catalysis [21]. Studies of solvent dependence in cases of problematic Sonogashira couplings are rather limited [2d, 22] as compared to the more common practice of an exhaustive ligand/catalyst search, in spite of a more practical change of solvent.
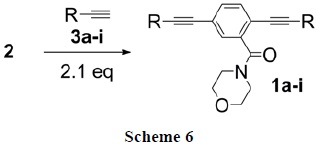
With reaction conditions established, a series of OPE3 products 1a-i were prepared in good yields (Scheme 6, Table 3). Despite the known acceleration of Sonogashira cross-couplings with alkynes substituted with electron withdrawing groups [23] couplings in DMSO worked equally well within 1 h with aryl acetylenes substituted either with strong electron donating (entry 5), electron withdrawing groups (entries 6-8) or heterocyclic acetylenes (entries 3 and 4). Compounds 1a-i were soluble in common organic solvents. 1H NMR spectra of all these compounds indicated high mobility of the morpholine moiety in solution.

At this stage it was decided to prepare OPE3s substituted with both electron donating groups and electron withdrawing groups in a push-pull electronic architecture, a common feature found in optoelectronic materials [3-5]. Thus, it was required to sequentially install different arylacetylene groups in dihaloarene 9 by means of a regioselective Sonogashira coupling at the more reactive C-I bond [1, 2d, 24]. The required dihaloarene 9 was in turn prepared by bromination [25] of acid 4 followed by treatment with thionyl chloride and morpholine (Scheme 7).

Sonogashira reactions of 9 in DMSO with aryl acetylenes 3a,f-h were carried out for preparation of mono coupled products 10a,f-h. (Scheme 8, Table 4). These reactions produced the desired mono coupled products 10a,f-h along with minor amounts of the undesired double coupled product 1a,f-h. Sonogashira coupling of 9 with arylacetylenes substituted with electron donating groups OCH3 (3a, entry 1) and NMe2 (3f, entry 2) clearly gave better results in terms of yield and selectivity than those observed using arylacetylenes bearing electron withdrawing groups CN (3g, entry 3) and NO2 (3h, entry 4).

Installation of the second arylacetylene group at the C-Br bond of compounds 10a,f-h proved more difficult than expected, but eventually was accomplished using reaction conditions reported by Buchwald [26] to afford compounds 1j-p. (Scheme 9) The low reactivity of substrates 10a and 10f (see Table 5) can be explained by the enhanced electron density at the C-Br bond due to the strong electron donating groups (OMe and NMe2) on the arylacetylene unit which likely made worse the Pd(0) oxidative addition step of the coupling reaction [1]. By contrast, bromoarene 10h bearing a nitro substituted arylacetylene (entry 7) gave compound 1p in higher yield.

With compounds 1a-p at hand, their photoluminescence properties were studied and results are shown in Table 6. Several compounds including the parent phenyleneethynylene 1b, methyl substituted 1e, and heterocyclic 3-pyridyl 1c and 3-thiophenyl analogs 1d display almost identical UV maxima absorbance. The rest of the compounds show bathochromic (red) shifts. All compounds are UV active and their ε values indicate intense absorption bands due to π-π* transitions. Compounds 1h, n, p containing one or two NO2 groups and the diketone compound 1i did not show fluorescence. All other compounds emit fluorescence in the 352-505 nm (violet to green) range. Compounds 1l and 1o, substituted with the electron withdrawing keto group, show modest fluorescence (entries 11 and 14).
Compounds with equal terminal aryl groups are listed in entries 1 through 9. Among them, compounds 1f with electron donating NMe2 substituents (entry 6) and 1g with electron withdrawing CN substituents (entry 7) displayed high quantum fluorescence yields, which were measured by Brouwer´s method [27] On the other hand, compounds with push-pull architecture (entries 10-15) show intramolecular charge transfer, as evidenced by large Stokes shifts which are associated to fluorescence quenching [28] However, cyano substituted compounds 1g (entry 7), 1k (entry 10) and 1m (entry 12) show high quantum fluorescence yields; the cyano group is a common feature of molecular wires [13b]. Similar OPE3s with high quantum yields are known [29]
In summary, we have prepared a series of soluble, fluorescent oligo phenyleneethynylenes OPE3, some of which display high quantum yields. Additionally, it was found that Sonogashira couplings proceeded much better in DMSO without need for other additives, the chemical yields were good and reactions took place within 1 h at 40-45°C. Couplings in DMSO containing 5% water gave a small increment of the yield of reactions of 2 with arylacetylenes substituted with electron donating groups.
Acknowledgments
Financial support from CONACYT-México (grants 61247 and 221360 and a predoctoral fellowship to MFJ) is gratefully acknowledged.
Experimental
General
Commercial reagents were used without further purification. Toluene was distilled from Na and stored over activated 4 Å molecular sieves under N2. THF was refluxed in the presence of triphenylphosphine then distilled and stored over activated 4-Å molecular sieves under N2. Reactions were monitored by TLC on Merck silica gel F254 plates and spots were visualized by a UV lamp at 254 and 365 nm. Column flash chromatography was performed using Whatman silica gel 60 (230-400 mesh). 1H and 13C NMR spectra were recorded on a Varian VNMR System 400 MHz spectrometer using tetramethylsilane (TMS δ=0.0 ppm) and CDCl3 (δ = 77.16 ppm) as internal standards; bs means broad signal, app d means apparent doublet in 1H NMR spectra. Infrared spectra were measured on a Perkin-Elmer FT-IR Spectrum GX spectrometer. High resolution mass spectra were recorded in a Jeol Gcmate mass spectrometer using EI (70 eV) as ionization mode. UV spectra were recorded on a Perkin-Elmer Lambda XLS spectrometer using quartz cells. For UV and fluorescence determinations spectroscopy quality CDCl3 was used. Fluorescence spectra were recorded at room temperature on a Perkin Elmer LS 55 spectrofluorometer. Fluorescence quantum yields φ in CHCl3 are relative to quinine sulfate 1N in H2SO4 [27]. Melting points were measured using a Büchi Melting Point B-540 apparatus and are uncorrected.
4-(2,5-Diiodobenzoyl)morpholine (2) Iodination of commercial 2-iodobenzoic acid 4 leading to 2,5-diiodobenzoic acid (5) was carried out according to a reported method [14]. Thus, in a 100 mL round bottom flask containing 30 mL of concentrated sulfuric acid were added 1.20 g of iodine and 0.34 g of NaIO4 at room temperature under vigorous magnetic stirring. When the solids dissolved it was added 2.48 g of 2-iodobenzoic acid 4 and the mixture was left stirring at room temperature (23°C) for 48 h. Then, the crude reaction mixture was poured on ice and the pink solid was filtered off. The solid was dissolved in ethyl acetate and washed with a saturated sodium thiosulfate solution. The organic layer was separated and dried with anhydrous sodium sulfate, filtered and concentrated in vacuo to afford 2.62 g (70%) of 5 as a white powder. Then, 1.68 g (4.5 mmol) of carboxylic acid 5 was suspended in 8 mL of thionyl chloride and heated to reflux for 2 h under N2 and allowed to cool to room temperature. Excess of thionyl chloride was removed in vacuo and the reaction mixture was diluted with 10 mL of anhydrous toluene. Catalytic DMAP and morpholine (1.6 mL, 18.0 mmol) were added under stirring at room temperature. After completion of the reaction (15 min), indicated by TLC analysis, the mixture was washed with 3% HCl solution, then with saturated NaHCO3 solution and extracted with ethyl acetate. The organic layer was dried with anhydrous Na2SO4 and concentrated to yield a yellow syrup that precipitated upon addition of hexane. Recrystallization from toluene-hexane afforded 1.40 g of 2 (70%) as an off-white powder, mp 145-146 °C; IR (KBr): λ 1600 (νN-C=O), 1700 (νc=0) cm-1; 1H NMR (400 MHz, CDCl3): δ 7.53 (d, J= 8.34, 1H), 7.50 (d, J= 1.99, 1H), 7.39 (dd, J=8.33, J=2.04, 1H), 3.9-3.1 (bs, 8H); 13C NMR (100 MHz, CDCl3): δ 167.8, 143.8, 140.9, 139.5, 135.9, 94.2, 91.8, 66.8, 66.6, 47.4, 42.1; HRMS (EI) m/z calcd. for C11H11I2NO2: 442.8880, found: 442.8886.
General procedure for double Sonogashira cross-coupling reactions of 4-(2,5-diiodobenzoyl)morpholine 2 with arylacetylenes 3a-i in DMSO
A mixture of 4-(2,5-diiodobenzoyl)morpholine 2 (177.2 mg, 0.40 mmol), alkynes 3a-i (0.84 mmol), Pd2(dba)3 (10.4 mg, 10 μmol), CuI (2.3 mg, 12 μmol), triphenylphosphine (6.3 mg, 24 μmol) and dry DMSO (5 mL) under N2, was degassed. iPr2NH (140 μL, 1 mmol) was added and then the mixture was heated at 45 °C (temperature of the external bath) under vigorous stirring. TLC analysis showed completion of the reaction after 1 h. After aqueous work-up and extraction with ethyl acetate the residue was adsorbed on silica gel and purified by flash chromatography (elution with hexane/EtOAc gradient). Evaporation of solvent gave a solid residue which was triturated with hexane/acetone to afford the desired pure product 1a-i.
The same procedure was followed for those reactions carried out in DMSO containing 5%, 33% and 67% of water (v/v) shown in Table 3.
2,5-Bis((4-ethoxyphenyl)ethynyl)morpholinebenzamide (1a) Obtained in 81% yield from the reaction of 2,5-diiodo-N-morpholinebenzamide (2) with 4-ethynylanisole (3a) as a white powder, mp 149-151 °C. IR (KBr,): λ 1604 (νN-C=O), 1633 (νc=0), 2213 (vCsp-Csp) cm-1; 1H NMR (400 MHz, CDCl3): δ 7.50-7.41 (m, 7H), 6.89 (app d, 4H), 3.90-3.25 (bs, m, 8H), 3.84 (s, 6H); 13C NMR (100 MHz, CDCl3): δ 168.3, 160.2, 160.1, 138.5, 133.32, 133.26, 132.0, 131.8, 129.6, 124.0, 119.9, 114.9, 114.7, 114.3, 114.2, 95.0, 92.4, 87.3, 85.6, 67.1, 67.0, 55.50, 55.48, 47.5, 42.3; HRMS (EI) m/z calcd. for C29H25NO4: 451.1784, found: 451.1765.
2,5-Bis(phenylethynyl)morpholinebenzamide (1b) Prepared in 60% yield by the reaction of 2,5-diiodo-N-morpholinebenzamide (2) with phenylacetylene (3b) as yellow syrup that solidifies upon standing. IR (film): λ 1599 (νNc=0), 1639 (νc=0), 2216 (vCsp-Csp) cm-1; 1H NMR (400 MHz, CDCl3): δ 7.55-7.47 (m, 7H), 7.38-7.34 (m, 6H), 3.95-3.20 (bs, m, 8H); 13C NMR (100 MHz, CDCl3): δ 168.0, 138.7, 132.2, 132.0, 131.8, 131.7, 129.7, 129.1, 128.9, 128.6, 128.5, 124.0, 122.7, 122.5, 119.8, 94.9, 92.4, 88.3, 86.6, 67.0, 66.9, 47.5, 42.2; HRMS (EI) m/z calcd. for C27H21NO2: 391.1572, found: 391.1589.
2,5-Bis(3-pyridinylethynyl)morpholinebenzamide (1c) Prepared in 86% yield by the reaction 2,5-diiodo-N-morpholinebenzamide (2) with 3-ethynylpyridine (3c) as an off-white powder, mp 157.7-158.2 °C. IR (KBr): λ 1613 (νc=0), 2211 (vCsp-Csp) cm-1 ; 1H NMR (400 MHz, CDCl3): δ 8.70 (bs, app d, 2H), 7.80 (app t, 3H), 7.61-7.51 (m, 4H), 7.33 (bs, s, 2H), 3.90-3.25 (bs, m, 8H); 13C NMR (100 MHz, CDCl3): δ 167.6, 152.4, 152.3, 149.4, 149.2, 139.0, 138.6, 138.5, 132.5, 132.1, 129.8, 123.8, 119.7, 123.25, 123.34, 91.5, 91.3, 89.6, 89.2, 67.0, 66.9, 47.5, 42.3; HRMS (EI) m/z calcd. for C25H19N3O2: 393.1477, found: 393.1482.
2,5-Bis(3-thiophenylethynyl)morpholinebenzamide (1d) Prepared in 90% yield by the reaction 2,5-diiodo-N-morpholinebenzamide (2) with 3-ethynylthiophene (3d) as a brown powder, mp 156.0-156.5 °C. IR (KBr): λ 1633 (νc=0), 2207 (vCsp-Csp) cm-1; 1H NMR (400 MHz, CDCl3): δ 7.55 (dd, J=2.97 Hz, J= 1.18 Hz, 1H), 7.53 (dd, J=2.98 Hz, J= 1.17 Hz, 1H), 7.51-7.49 (m, 2H), 7.49-7.48 (app d, 1H), 7.33 (t, J= 2.93, 1H), 7.32 (t, J= 2.93, 1H) , 7.19 (dd, J=5.02 Hz, J= 1.18 Hz, 1H), 7.16 (dd, J=4.97 Hz, J= 1.18 Hz, 1H), 3.90-3.25 (bs, m, 8H); 13C NMR (100 MHz, CDCl3): δ 167.9, 138.5, 132.0, 131.8, 129.7, 129.6, 129.53, 129.46, 129.3, 125.6, 123.8, 121.7, 121.5, 119.6, 89.9, 87.8, 87.5, 86.0, 66.9, 66.8, 47.3, 42.1; HRMS (EI) m/z calcd. for C23H17NO2S2: 403.0701, found: 403.0706.
2,5-Bis(p-tolylethynyl)morpholinebenzamide (1e) Prepared in 70% yield by the reaction 2,5-diiodo-N-morpholinebenzamide (2) with 4-methylphenylacetylene (3e) as a white powder, mp 179.9-180.8 °C. IR (KBr): λ 1630 (νc=0), 2213 (vCsp-Csp) cm-1; 1H NMR (400 MHz, CDCl3): δ 7.52-7.48 (m, 3H), 7.42 (app d, 2H), 7.38 (app d, 2H), 7.17 (app d, 4H), 3.95-3.20 (bs, m, 8H), 2.38 (s, 6H); 13C NMR (100 MHz, CDCl3): δ 168.0, 139.2, 139.0, 138.5, 132.0, 131.8, 131.5, 131.5, 129.6, 129.3, 129.2, 123.9, 119.7, 119.5, 119.3, 95.0, 92.5, 87.7, 85.9, 66.9, 66.8, 47.3, 42.1; HRMS (EI) m/z calcd. for C29H25NO2: 419.1885, found: 419.1890.
2,5-Bis((4-(dimethylamino)phenyl)ethynyl)morpholinebenzamide (1f) Prepared in 83% yield by the reaction 2,5-diiodo-N-morpholinebenzamide (2) with 4-ethynyl-N,N-dimethylaniline (3f) as a yellow powder, mp 209.4-210.2 °C. IR (KBr): λ 1607(nN-C=O), 1630 (νc=0), 2204 (vCsp-Csp) cm-1; 1H NMR (400 MHz, CDCl3): δ 7.45 (s, 3H), 7.41-7.34 (m, 4H), 6.68-6.62 (m, 4H), 3.92-3.20 (bs, m, 8H), 3.00 (s, 12H); 13C NMR (100 MHz, CDCl3): δ 168.6, 150.5, 150.4, 138.1, 133.0, 132.9, 131.7, 131.6, 129.4, 124.0, 119.9, 111.91, 111.89, 109.5, 109.2, 96.3, 93.6, 86.9, 85.2, 67.1, 67.0, 47.5, 42.2, 40.33, 40.31; HRMS (EI) m/z calcd. for C31H31N3O2: 477.2416, found: 477.2432.
2,5-Bis((4-cyanophenyl)ethynyl)morpholinebenzamide (1g) Prepared in 80% by the reaction 2,5-diiodo-N-morpholinebenzamide (2) with 4-ethynylbenzonitrile (3g) as a yellow powder, mp 228.0-230.0 °C (dec.) IR (KBr): λ 1602 (nN-C=O), 1640 (νc=0), 2217 (vCsp-Csp), 2227 (νCsp-N) cm-1; 1H NMR (400 MHz, CDCl3): δ 7.68 (app dd, 4H), 7.64-7.54 (m, 7H), 3.9-3.25 (bs, m, 8H); 13C NMR (100 MHz, CDCl3): δ 167.3, 139.1, 132.5, 132.3, 132.3, 132.2, 132.1, 129.9, 127.3, 127.0, 123.7, 119.6, 118.3, 118.2, 112.4, 112.1, 93.1, 92.0, 90.9, 90.2, 66.9, 66.8, 47.4, 42.2; HRMS (EI) m/z calcd. for C29H19N3O2: 441.1477, found: 441.1488.
2,5-Bis((4-nitrophenyl)ethynyl)morpholinebenzamide (1h) Prepared in 80% yield by the reaction 2,5-diiodo-N-morpholinebenzamide (2) with 1-ethynyl-4-nitrobenzene (3h) as a yellow powder, mp 204.8-205.7 °C. IR (KBr): λ 1347 and 1519 (νNO2), 1593 (νN-C=O), 1638 (νc=0), 2218 (vCsp-Csp), cm-1; 1H NMR (400 MHz, CDCl3): δ 8.26 (app d, 4H), 7.71-7.55 (m, 7H), 3.90-3.25 (bs, m, 8H); 13C NMR (100 MHz, CDCl3): δ 167.4, 147.6, 147.4, 139.3, 132.7, 132.6, 132.5, 132.3, 130.0, 129.4, 129.0, 124.0, 123.8, 119.7, 93.0, 92.9, 91.1, 90.8, 67.0, 66.9, 47.5, 42.3; HRMS (EI) m/z calcd. for C27H19N3O6: 481.1274, found: 481.1280.
2,5-Bis((4-acetophenyl)ethynyl)morpholinebenzamide (1i) Prepared in 71% yield by the reaction 2,5-diiodo-N-morpholinebenzamide (2) with 4-ethynylacetophenone (3i) as a green-yellow powder, mp 194.1-196.0 °C. IR (KBr): 1600 (νN-C=O), 1630 (νc=0), 1677 (νc=0), 1690 (νc=0), 2208 (vCsp-Csp) cm-1; 1H NMR (400 MHz, CDCl3): δ 7.95 (app d, 4H), 7.63-7.52 (m, 7H), 3.90-3.25 (bs, m, 8H), 2.61 (s, 6H); 13C NNMR (100 MHz, CDCl3): δ 197.4, 197.3, 167.7139.1, 136.9, 136.7, 132.5, 132.2, 131.9, 131.9, 129.9, 128.6, 128.5, 127.4, 127.1, 123.8, 119.8, 94.2, 91.8, 91.2, 89.5, 67.0, 66.9, 47.5, 42.3, 26.8; HRMS (EI) m/z calcd. for C31H25NO4: 475.1784, found: 475.1800.
4-(5-Bromo-2-iodobenzoyl)morpholine (9) Bromination of commercial 2-iodobenzoic acid 4 leading to 5-bromo-2-iodobenzoic acid (8) was carried out according to a reported method [25] Thus, in a 100 mL round bottom flask containing 30 mL of concentrated sulfuric acid at 60 °C, was added 2-iodobenzoic acid 4 (3.72 g, 15 mmol), then maintaining this temperature three portions of solid NBS (6 mmol) were added every 15 min, for a total of 3.2 g (18 mmol) of NBS. After the last addition, the mixture was left stirring for 1 h at 60°C, then left to cool to room temperature and poured over crushed ice. The pink solid was filtered off and dissolved in ethyl acetate and washed with a concentrated solution of sodium thiosulfate. The organic portion was dried with anhydrous Na2CO3, filtered and concentrated in vacuo to afford 3.58 g (73%) of the carboxylic acid 8 as a white solid. Then, 1.471 g (4.5 mmol) of carboxylic acid 8 was suspended in 8 mL of thionyl chloride and heated to reflux for 2 h under N2 and allowed to cool to room temperature. Excess of thionyl chloride was removed in vacuo and the reaction mixture was diluted with 10 mL of anhydrous toluene. Catalytic DMAP and morpholine (1.6 mL, 18.0 mmol) were added under stirring at room temperature. After completion of the reaction (15 min), indicated by TLC analysis, the mixture was washed with 3% HCl solution, then with saturated NaHCO3 and extracted with ethyl acetate. The organic layer was dried with anhydrous Na2SO4 and concentrated to yield a yellow syrup that precipitated upon addition of hexane. Recrystallization from toluene-hexane afforded 1.301 g of 10 (73%) as a white powder, mp 148 °C; IR (KBr) λ 1620 (νN-C=O) cm-1, 1H NMR (400 MHz, CDCl3): δ 7.68 (d, J= 8.44, 1H), 7.34 (d, J= 2.31, 1H), 7.22 (dd, J=8.43, J=2.35, 1H), 3.9-3.1 (bs, 8H); 13C NMR (100 MHz, CDCl3): δ167.92, 143.60, 140.76, 133.67, 130.15, 123.09, 90.55, 66.76, 66.62, 47.31, 42.12; HRMS (EI) m/z calcd. for C11H11BrINO2: 394.9018, found: 394.9020
General procedure for C-I regioselective Sonogashira cross-coupling reactions of 9 with arylacetylenes. A mixture of 9 (158.4 mg, 0.40 mmol), alkynes 3a,f-h (0.44 mmol), Pd2(dba)3 (10.4 mg, 10 μmol), CuI (2.3 mg, 12 μmol), triphenylphosphine (6.3 mg, 24 mmol) and DMSO (5 mL) under N2, was degassed. iPr2NH (140 μL, 1 mmol) was added and then the mixture was heated at 45 °C (temperature of external bath) under vigorous stirring for 1 h as TLC analysis indicated completion of the reaction. Aqueous work-up and extraction gave the crude reaction mixture which was adsorbed on silica gel and after flash chromatography (elution with hexane/EtOAc gradient) the solid residue was washed with hexane/acetone to afford the desired pure products 10a, f-h.
5-Bromo-2-((4-methoxyphenyl)ethynyl)morpholinebenzamide (10a) Prepared in 73% yield by the reaction of 5-bromo-2-iodo-N-morpholinebenzamide (9) with 4-ethynylanisole (6a) as a yellow powder, mp 138-139 °C. IR (KBr): λ 1600 (νN-C=O), 1692 (νc=0), 2212 (νCsp- Csp) cm-1; 1H NMR (400 MHz, CDCl3): δ 7.52-7.47 (m, 2H), 7.44-7.37 (m, 3H), 6.88 (app d, 2H), 3.90-3.25 (bs, m, 8H), 3.84 (s, 3H); 13C NMR (100 MHz, CDCl3): δ 167.2, 160.1, 139.7, 133.2, 133.1, 132.2, 129.7, 122.5, 119.5, 114.24, 114.19, 94.4, 84.6, 66.8, 66.7, 55.4, 47.3, 42.1; HRMS (EI) m/z calcd. for C20H18BrNO3: 399.0470, found: 399.0472.
5-Bromo-2-((4-(dimethylamino)phenyl)ethynyl)morpholinebenzamide (10f) Prepared in 72% yield by the reaction of 5-bromo-2-iodo-N-morpholinebenzamide (9) with 4-ethynyl-N, N-dimethylaniline (3f) as a yellow powder, mp 123-124 °C. IR (KBr): λ 1604 (νN-C=O), 1688(νc=0), 2204 (νCsp- Csp) cm-1; 1H NMR (400 MHz, CDCl3): δ 7.49-7.45 (m, 2H), 7.38-7.32 (m, 3H), 6.64 (app d, 2H), 3.90-3.25 (bs, m, 8H), 3.00 (s, 6H); 13C NMR (100 MHz, CDCl3): d167.4, 150.4, 139.4, 133.0, 132.8, 132.1, 129.7, 121.7, 120.1, 111.7, 108.6, 96.0, 84.0, 66.8, 66.7, 47.3, 42.1, 40.1; HRMS (EI) m/z calcd. for C21H21BrN2O2: 412.0786, found: 412.0770.
5-Bromo-2-((4-cyanophenyl)ethynyl)morpholinebenzamide (10g) Prepared in 43% yield by the reaction of 5-bromo-2-iodo-N-morpholinebenzamide (9) and 4-ethynylbenzonitrile (3g) as a yellow powder, mp 237-238 °C (dec). IR (KBr): λ 1602 (νN-C=O), 1687 (νc=0), 2224 (νCsp-N) cm-1; 1H NMR (400 MHz, CDCl3): δ 7.66 (app d, 2H), 7.58-7.54 (m, 3H), 7.51 (app d, 1H), 7.44(dd, J=8.2 Hz J= 0.26 Hz, 1H), 3.90-3.25 (bs, m, 8H); 13C NMR (100 MHz, CDCl3): δ 166.9, 140.4, 133.8, 132.6, 132.4, 132.2, 130.0, 127.2, 124.2, 118.4, 118.3, 112.5, 92.2, 89.9, 67.0, 66.9, 47.5, 42.3; HRMS (EI) m/z calcd. for C20H15BrN2O2: 394.0317, found: 394.0306.
5-Bromo-2-((4-nitrophenyl)ethynyl)morpholinebenzamide (10h) Prepared in 42% yield by the reaction of 5-bromo-2-iodo-N-morpholinebenzamide (9) with 1-ethynyl-4-nitrobenzene (3h) as a yellow powder, mp 229-230 °C (dec). IR (KBr): λ 1347 and 1528 (νNO2), 1600 (νN-C=O), 1691 (νc=0), 2217 (νCsp- Csp) cm-1; 1H NMR (400 MHz, CDCl3): δ 8.23 (app d, 2H), 7.62 (app d, 2H), 7.56 (dd, J=8.30 Hz J= 2.01 Hz, 1H), 7.52 (d, J= 1.97 Hz, 1H), 7.45 (d, J= 8.27 Hz, 1H), 3.90-3.25 (bs, m, 8H); 13C NMR (100 MHz, CDCl3): δ 166.7, 147.4, 140.3, 133.7, 132.5, 132.3, 129.8, 129.0, 124.2, 123.8, 118.0, 91.8, 90.6, 66.8, 66.8, 47.4, 42.2; HRMS (EI) m/z calcd. for C19H15BrN2O4: 414.0215, found: 414.0227.
General procedure for Sonogashira cross-coupling reactions used to prepare 1j-p. Compounds 10a, 10f and 10h were subjected to Sonogashira couplings at the C-Br bond with arylacetylenes following conditions reported by Buchwald.26 Thus, a mixture of the bromoarene 10a,f-h (0.40 mmol, 1.0 equiv), arylalkynes 3a,g-1 (0.44 mmol, 1.1 equiv), PdCl2(CH3CN)2 (3.1 mg, 0.03 equiv, 12 mmol), XPhos (11.4 mg, 24 mmol, 0.06 equiv), Cs2CO3 (338.9 mg, 1.04 mmol, 2.6 equiv) and CH3CN (6 mL) under N2, was degassed. Then, the mixture was heated at 75 °C (temperature of external bath) under vigorous stirring for 12 h. After this time, TLC analysis of the reaction mixture indicated completion of the reaction. Aqueous work-up and extraction with ethyl acetate (3 × 15 mL) gave the crude reaction mixture which was dried over anhydrous Na2CO3, filtered, and finally adsorbed on silica gel. After flash chromatography (elution with hexane/EtOAc gradient) the solid residue was washed with hexane/acetone to afford the desired pure products 1j-p.
5-((4-Cyanophenyl)ethynyl)-2-((4-methoxyphenyl)ethynyl)morpholinebenzamide (1j) Prepared in 80% yield by the reaction of 10a with 4-ethynylbenzonitrile (3g) as a yellow powder, mp 188.4-188.8 °C (dec). IR (KBr): λ 1605 (νN-C=O), 1625 (νc=0), 2205 (vCsp-Csp), 2225 (νCsp-N) cm-1; 1H NMR (400 MHz, CDCl3): δ 7.66 (app d, 2H), 7.60 (app d, 2H), 7.54-7.52 (m, 3H), 7.44 (app d, 2H), 6.90 (app d, 2H), 3.90-3.25 (bs, m, 8H), 3.85 (s, 3H); 13C NMR (100 MHz, CDCl3): δ 167.8, 160.2, 138.5, 133.2, 132.1, 131.98, 132.02, 132.0, 129.9, 127.6, 122.3, 121.1, 118.4, 114.2, 114.2, 111.9, 95.7, 92.4, 90.2, 85.2, 66.9, 66.8, 55.4, 47.3, 42.1; HRMS (EI) m/z calcd. for C29H22N2O3: 446.1631, found: 446.1627.
5-((4-Acetophenyl)ethynyl)-2((4-methoxyphenyl)ethynyl)morpholinebenzamide (1l) Prepared in 55% yield from the reaction of 10a with 4-ethynylacetophenone (3i) as a yellow powder, mp 182.0-182.5 °C. IR (KBr): λ 1605 (νN-C=O), 1627 (νc=0), 1677 (νc=0), 2208 (vCsp-Csp) cm-1; 1H NMR (400 MHz, CDCl3): δ 7.96 (app d, 2H), 7.60 (app d, 2H), 7.53 (s, 3H), 7.44 (app d, 2H), 6.90 (app d, 2H), 3.90-3.25 (bs, m, 8H), 3.85 (s, 3H), 2.63 (s, 3H); 13C NMR (100 MHz, CDCl3): δ 197.2, 167.9, 160.2, 138.5, 136.4, 133.2, 132.0, 131.9, 131.7, 129.8, 128.3, 127.5, 122.8, 120.8, 114.3, 114.2, 95.5, 91.4, 91.2, 85.3, 66.9, 66.8, 55.3, 47.3, 42.1, 26.7; HRMS (EI) m/z calcd. for C30H25NO4: 463.1784, found: 463.1804.
5-((4-Cyanophenyl)ethynyl)-2-((4-(dimethylamino)phenyl)ethynyl)morpholine benzamide (1m) Prepared in 36% yield by the reaction of 10f with 4-ethynylbenzonitrile (3g) as a yellow powder, mp 210.7-212.1 °C. IR (KBr): λ 1593 (νN-C=O), 1633 (νc=0), 2213 (vCsp-Csp), 2223 (νCsp-N) cm-1; 1H NMR (400 MHz, CDCl3): δ 7.65 (app d, 2H), 7.59 (app d, 2H), 7.50 (s, 3H), 7.36 (d app, 2H), 6.65 (app d, 2H), 3.95-3.20 (bs, m, 8H), 3.01 (s, 6H); 13C NMR (100 MHz, CDCl3): δ 168.0, 150.5, 138.1, 132.9, 132.09, 132.07, 132.0, 131.7, 130.0, 127.7, 121.8, 121.6, 118.5, 111.7, 111.7h, 108.5, 97.5, 92.7, 89.9, 84.8, 66.9, 66.8, 47.3, 42.1, 40.1; HRMS (EI) m/z calcd. for C30H25N3O2: 459.1947, found: 459.1968.
2-((4-(Dimethylamino)phenyl)ethynyl)-5-((4-nitrophenyl)ethynyl)morpholine benzamide (1n) Prepared in 35% yield by the reaction of 10f with 1-ethynyl-4-nitro benzene (3h) as a red powder, mp 227.3 - 227.9 °C. IR (KBr): λ 1339 and 1512 (νNO2), 1589 (νN-C=O), 1624 (νc=0), 2205 (vCsp-Csp) cm-1; 1H NMR (400 MHz, CDCl3): δ 8.21 (app d, 2H), 7.64 (app d, 2H), 7.53-7.49 (m, 3H), 7.36 (app d, 2H), 6.64 (app d, 2H), 3.95-3.20 (bs, m, 8H), 3.00 (s, 6H); 13C NMR (100 MHz, CDCl3): δ 167.9, 150.5, 147.1, 138.2, 132.9, 132.3, 132.0, 131.7, 130.0, 129.7, 123.7, 122.0, 121.4, 111.7, 108.5, 97.6, 93.6, 89.7, 84.8, 66.9, 66.8, 47.4, 42.1, 40.1; HRMS (EI) m/z calcd. for C29H25N3O4: 479.1845, found: 479.1832.
5-((4-Acetophenyl)ethynyl)-2-((4-(dimethylamino)phenyl)ethynyl)morpholine benzamide (1o) Prepared in 35% yield by the reaction of 10f with 4-ethynylacetophenone (3i) as a yellow powder, mp 214.8-215.3 °C. IR (KBr): λ 1592 (νN-C=O), 1641 (νc=0), 1677 (νc=0), 2199 (vCsp-Csp) cm-1; 1H NMR (400 MHz, CDCl3): δ 7.94 (app d, 2H), 7.59 (app d, 2H), 7.52-7.47 (m, 3H), 7.35 (app d, 2H), 6.64 (app d, 2H), 3.95-3.20 (bs, m, 8H), 2.99 (s, 6H), 2.60 (s, 3H); 13C NMR (100 MHz, CDCl3): δ 197.3, 168.0, 150.5, 138.1, 136.4, 132.9, 131.9, 131.7, 131.6, 129.9, 128.3, 127.6, 122.0, 121.4, 111.7, 108.6, 97.2, 91.7, 90.9, 84.8, 66.9, 66.8, 47.3, 42.1, 40.1, 26.7; HRMS (EI) m/z calcd. for C31H28N2O3: 476.2100, found: 476.2130.
5-((4-Methoxyphenyl)ethynyl)-2-((4-nitrophenyl)ethynyl)morpholinebenzamide (1p) Prepared in 65% yield by the reaction of 10h and 4-ethynylanisole (3a) as a yellow powder, mp 194.3-194.5 °C. IR (KBr): λ 1340 y 1513 (νNO2), 1590 (νN-C=O), 1636 (νc=0), 2209 (vCsp-Csp), cm-1; 1H NMR (400 MHz, CDCl3): δ 8.24 (app d, 2H), 7.63 (app d, 2H), 7.58-7.50 (m, 3H), 7.47 (app d, 2H), 6.90 (app d, 2H), 3.90-3.25 (bs, m, 8H), 3.84 (s, 3H); 13C NMR (100 MHz, CDCl3): δ 167.8, 160.3, 147.4, 139.1, 133.4, 132.6, 132.4, 131.9, 129.5, 129.4, 125.6, 124.0, 118.2, 114.5, 114.3, 93.5, 92.4, 91.7, 87.0, 67.1, 67.0, 55.5, 47.5, 42.3; HRMS (EI) m/z calcd. for C28H22N2O5: 466.1529, found: 466.1512.
References
1. a) Sonogashira, K. J. Organomet. Chem. 2002, 653, 46-49. [ Links ] b) Chinchilla, R.; Nájera, C. Chem. Soc. Rev., 2011, 40, 5084-5121. [ Links ] c) Chinchilla, R.; Nájera, C. Chem. Rev. 2007, 107, 874-922. [ Links ]
2. a) Cassar L. J. Organomet. Chem. 1975, 93, 253-257. [ Links ] b) Dieck, H. A.; Heck, F. R. J. Organomet. Chem. 1975, 93, 259-263. [ Links ] c) Alami, M.; Ferri, F.; Linstrumelle, G. Tetrahedron Lett. 1993, 34, 6403-6406. [ Links ] d) Doucet, H.; Hierso, J. C. Angew. Chem. Int. Ed. 2007, 46, 834-871. [ Links ] e) Tykwinski, R. R. Angew. Chem. Int. Ed. 2003, 42, 1566-1568. [ Links ] f) Plenio, H. Angew. Chem. Int. Ed. 2008, 47, 6954-6956. [ Links ]
3. a) Bunz, U. H. F. Chem. Rev. 2000, 100, 1605-1644. [ Links ] b) Bunz, U. H. F. Adv. Poly. Sci. 2005, 177, 1-52. [ Links ]
4. Yamamoto, T.; Yamaguchi, I.; Yasuda, T. Adv. Polym. Sci. 2005, 177, 181-208. [ Links ]
5. Klemm, E.; Pautzsch, T.; Blankenburg, L. Adv. Polym. Sci. 2005, 177, 53-90. [ Links ]
6. a) Tour J. M. Acc. Chem. Res. 2000, 33, 791-804. [ Links ] b) Voskerician, G.; Weder, C. Adv. Poly. Sci. 2005, 177, 209-248. [ Links ] c) Liu, J.; Lam, J. W. Y.; Tang, B. Z. Chem. Rev. 2009, 109, 5799-5867. [ Links ] d) Swager, T. M.; Gil, C. J.; Wrighton M. S. J. Phys. Chem. 1995, 99, 4886-4893. [ Links ] Vokatáand, T.; Moon J. H. Macromolecules, 2013, 46, 1253-1259. [ Links ]
7. James, D. K.; Tour, J. M. Top. Curr. Chem. 2005, 257, 33-62. [ Links ]
8. a) Swager, T. M.; Zheng, J. Adv. Polym. Sci. 2005, 177, 151-179. [ Links ] b) Swager, T. M. Acc. Chem. Res. 1998, 31, 201-207. [ Links ] c) McQuade, D. T.; Pullen, A. E.; Swager T. M. Chem. Rev., 2000, 100, 2537-2574. [ Links ]
9. a) Miranda, O. R.; You, C. C.; Phillips, R.; Kim, I. B.; Ghosh, P. S.; Bunz, U. H.; Rotello, V. M. J. Am. Chem. Soc. 2007, 129, 9856-9857. [ Links ] b) You, C. C.; Miranda, O. R.; Gider, B.; Ghosh, P. S.; Kim, I. B.; Erdogan, B.; Krovi, S. A.; Bunz, U. H.; Rotello, V. M. Nat. Nanotechnol. 2007, 2, 318-323. [ Links ] c) Bajaj, A.; Miranda, O. R.; Kim, I. B.; Phillips, R. L.; Jerry, D. J.; Bunz, U. H.; Rotello, V. M. Proc. Natl. Acad. Sci. U.S.A. 2009, 106, 10912-10916. [ Links ] d) Zhu, C.; Liu, L.; Yang, Q.; Lv, F.; Wang, S. Chem. Rev. 2012, 112, 4687-4735. [ Links ] e) Vendrell, M.; Zhai, D.; Er, J. C.; Chang, Y. T. Chem. Rev. 2012, 112, 4391-4420. [ Links ]
10. Voskerician, G.; Weder, C. Adv. Polym. Sci. 2005, 177, 209-248. [ Links ]
11. a) Tour, J. M. Chem. Rev. 1996, 96, 537-554. [ Links ] b) Xue, C.; Luo, F. T. Tetrahedron, 2004, 60, 6285–6294. [ Links ] c) Wang, C.; Batsanov, A. S.; Bryce, M. R.; Sage, I. Org. Lett. 2004, 6, 2181-2184. [ Links ] d) Wang, C.; Batsanov, A. S.; Bryce, M. R. J. Org. Chem. 2006, 71, 108-116. [ Links ] e) Mayr, A.; Srisailas, M.; Zhao, Q.; Gao, Y.; Hsieh, H.; Hoshmand-Kochi, M.; St. Fleur, N. Tetrahedron, 2007, 63, 8206-8217. [ Links ] f) Wang, L. G.; Zhan, T. G.; Zhao, X.; Jiang, X. K.; Li, Z. T. Tetrahedron, 2012, 68, 5303-5310. [ Links ] g) Bai, X.; Chen, X.; Barnes, C.; Dias, J. R.; Sandreczki, T. C. Tetrahedron, 2013, 69, 1105-1111. [ Links ]
12. a) Dirk, S. M.; Tour, J. M. Tetrahedron, 2003, 59, 287-293. [ Links ] b) Price, D. W.; Tour, J. M. Tetrahedron, 2003, 59, 3131-3156. [ Links ] c) Price, D. W.; Dirk, S. M.; Maya, F.; Tour, J. M. Tetrahedron, 2003, 59, 2497-2518. [ Links ] d) Neto, B. A. D.; Lopes, A. S.; Ebeling, G.; Goncalves, R. S.; Costa, V. E. U.; Quinab, F. H.; Dupont, J. Tetrahedron, 2005, 61, 10975-10982. [ Links ] e) Stuhr-Hansen, N.; Sørensen, J. K.; Moth-Poulsen, K.; Christensen, J. B.; Bjørnholm, T.; Nielsen, M. B. Tetrahedron 2005, 61, 12288-12295. [ Links ] f) Dixon, S.; Whitby, R. J. Tetrahedron Letters, 2006, 47, 8147-8150. [ Links ] g) Chandra, K. L.; Zhang, S.; Gorman, C. B. Tetrahedron, 2007, 63, 7120-7132. [ Links ] h) Ryhding, T.; Kirketerp, M. B. S.; Kadhane, U.; Lykkegaard, M. K.; Panja, S.; Nielsen, S. B.; Nielsen, M. B. Tetrahedron, 2008, 64, 11475-11479. [ Links ]
13. a) Beeby, A.; Findlay, K.; Low, P. J.; Marder, T, B. J. Am. Chem. Soc. 2002, 124, 8280-8284. [ Links ] b) Dirk, S. M.; Tour, J. M. Tetrahedron 2003, 59, 287-293. [ Links ]
14. a) Kraszkiewicz, L.; Sosnowski, M.; Skulski L. Tetrahedron 2004, 60, 9113-9119. [ Links ] b) Kraszkiewicz, L.; Sosnowski, M.; Skulski L. Synthesis 2006, 1195-1199. [ Links ]
15. a) Reichardt, C. Chem. Rev. 1994, 94, 2319-2358. [ Links ] b) Reichardt, C.; Schäfer, G. Liebigs Ann. 1995, 1579-1582. [ Links ] c) Eberhardt, R.; Löbbecke, S.; Neidhardt, B.; Reichardt, C. Liebigs Ann./Recueil. 1997, 1195-1199. [ Links ] d) Reichardt, C. Green Chem. 2005, 7, 339-351. [ Links ]
16. Wagner, R. W.; Johnson, T. E.; Li, F.; Lindsey, J. S. J. Org. Chem. 1995, 60, 5266-5273. [ Links ]
17. Christiansen, E.; Due-Hansen, M. E.; Ulven, T. J. Org. Chem. 2010, 75, 1301-1304. [ Links ]
18. Catalán, J.; Díaz, C.; García-Blanco, F. J. Org. Chem. 2001, 66, 5846-5852. [ Links ]
19. Diao, T.; White, P.; Guzei, I.; Stahl, S. Inorg. Chem. 2012, 51, 11898-11909. [ Links ]
20. a) Lee, H. B.; Huh, D. H.; Oh, J. S.; Min, G. H.; Kim, B. H.; Lee, D. H.; Hwang, J. K.; Kim, Y. G. Tetrahedron. 2001, 57, 8283-8290. [ Links ] b) Köllhofer A.; Plenio H. Chem. Eur. J. 2003, 9, 1416-1425. [ Links ] c) Remmele, H.; Köllhofer, A.; Plenio, H. Organometallics 2003, 22, 4098-4103. [ Links ] d) Shirakawa, E.; Kitabata, T.; Otsuka, H.; Tsuchimoto, T. Tetrahedron 2005, 61, 9878-9885. [ Links ] e) Yi C.; Hua R. J. Org. Chem. 2006, 71, 2535-2537. [ Links ] f) Huang M.; Feng Y.; Wu, Y. Tetrahedron 2012, 68, 376-381. [ Links ] g) Gallop, C. W. D.; Chen, M. T.; Navarro, O. Org. Lett. 2014, 16, 3724-3727. [ Links ]
21. a) Grennberg, H.; Gogoll, A.; Bäckvall, J. E. J. Org. Chem. 1991, 56, 5808-5811. [ Links ] b) Steinhoff, B. A.; Stahl, S. S. J. Am. Chem. Soc. 2006, 128, 4348-4355. [ Links ] c) Vasseur, A.; Harakat D.; Muzart, J.; Le Bras, J. J. Org. Chem. 2012, 77, 5751-5758. [ Links ] d) Qi, C. Z.; Sun, X. D.; Lu, C. Y; Yang, J. Z.; Du, Y. J.; Wu, H. J.; Zhang, X. M. J. Organomet. Chem. 2009, 694, 2912-2916. [ Links ] e) Zierkiewicz, W.; Privalov, T. Organometallics. 2005, 24, 6019-6028. [ Links ]
22. Ljungdahl, T.; Pettersson, K.; Albinsson, B., Mårtensson, J. J. Org. Chem. 2006, 71, 1677-1687. [ Links ]
23. Schilz, M.; Plenio, H. J. Org. Chem. 2012, 77, 2798-2807. [ Links ]
24. Mphahlele M. J.; Lesenyeho L. G.; Makelane H. R. Tetrahedron, 2010, 66, 6040-6046. [ Links ]
25. Rajesh, K.; Somasundaram, M.; Saiganesh, R.; Balasubramanian, K. K. J. Org. Chem. 2007, 72, 5867-5869. [ Links ]
26. Gelman, D.; Buchwald, S. L. Angew. Chem. Int. Ed. 2003, 42, 5993-5996. [ Links ]
27. Brouwer, A. M. Pure Appl. Chem. 2011, 83, 2213-2228. [ Links ]
28. Tonzola, C. J.; Alam, M. M.; Kaminsky, W.; Jenekhe, S. A. J. Am. Chem. Soc. 2003, 125, 13548-13558. [ Links ]
29. a) Bai, X.; Chen, X.; Dias, J. R.; Sandreczki T. C. Tetrahedron Lett. 2013, 54, 1711-1713. [ Links ] b) Fang, J. K.; An, D. L.; Wakamatsu, K.; Ishikawa, T.; Iwanaga, T.; Toyota, S.; Akita, S.; Matsuo, D.; Orita, A.; Otera, J. Tetrahedron. 2010, 66, 5479-5485. [ Links ]

