










Servicios Personalizados
Revista
Articulo
 Inglés (pdf)
Inglés (pdf)
 Artículo en XML
Artículo en XML Referencias del artículo
Referencias del artículo
 Enviar artículo por email
Enviar artículo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO -
 Accesos
Accesos
Links relacionados
-
 Similares en
SciELO
Similares en
SciELO
Compartir

 Permalink
PermalinkJournal of the Mexican Chemical Society
versión impresa ISSN 1870-249X
J. Mex. Chem. Soc vol.59 no.2 Ciudad de México abr./jun. 2015
Articles
ZnO-Photocatalyzed Oxidative Transformation of Diphenylamine. Synergism by TiO2, V2O5, CeO2 and ZnS
Chockalingam Karunakaran* and Swaminathan Karuthapandian1
* Corresponding author: Prof. Dr. C. Karunakaran, CSIR Emeritus Scientist, Department of Chemistry, Annamalai University, Annamalainagar 608002, India.
1 Present address: Department of Chemistry, VHNSN College, Virudhunagar 626001, Tamilnadu, India.
Received September 24th, 2014
Accepted February 17th, 2015
Abstract
Diphenylamine (DPA) undergoes photocatalytic transformation on nanoparticulate ZnO surface yielding N-phenyl-p-benzoquinonimine (PBQ). The reaction rate increases with the increase of (i) DPA-concentration, (ii) ZnO-loading, (iii) airflow rate and (iv) light intensity. The formation of PBQ is larger on using UV-C light instead of UV-A light. The photocatalyst is reusable. The mechanism of the photocatalytic transformation has been discussed with a suitable kinetic law. Nanoparticulate TiO2, V2O5, CeO2 and ZnS enhance the ZnO-photocatalyzed PBQ formation indicating interparticle charge transfer in semiconductor mixtures.
Key words: Photocatalysis; Semiconductor; Kinetic law; Interparticle charge transfer; N-phenyl-p-benzoquinonimine.
Resumen
La Difenil amina (DPA) se transforma en N-fenil-p-benzoquinonimina (PBQ) a través de un proceso fotocatalítico en la superficie de ZnO nanoestructurado. La velocidad de reacción aumenta con (i) la concentración DPA, (ii) la cantidad de ZnO, (iii) el flujo de aire y, (iv) la intensidad de la luz. La tasa de de formación de PBQ es mayor si se usa iluminación con UV-C comparada con UV-A. El fotocatalizador se puede usar de nuevo. El mecanismo de la conversión fotocatalítica se discute en términos de una ley cinética apropiada. La presencia de TiO2, V2O5, CeO2 y ZnS nanoestructurados incrementa la formación fotocatalítica de PBQ en ZnO nanoestructurado, indicando que hay transferencia de carga entre las partículas de los diferentes materiales semiconductores.
Palabras clave: Fotocatalísis; Semiconductor; Ley cinética; Transferencia de carga entre partículas; N-fenil-p-benzoquinonimina.
Introduction
Heterogeneous photocatalysis provides a promising method to realize green chemical process in organic synthesis and application of semiconductor-photocatalysis in selective organic transformations has been highlighted by Lang et al [1], Palmisano et al [2], Shiraishi and Hirai [3], etc. Selective photocatalytic (i) oxidation of alcohols to aldehydes by TiO2 [1, 3, 4], CdS [5] and Pd@CdS [6] and to ketones by TiO2 [2] and Au/TiO2 [1], benzene to phenol by Au/TiO2 [1], amines to imines by TiO2 [1], Pt/TiO2 [3] and Au/TiO2 [1], (ii) oxidative dehydrogenation of 1,2,3,4-tetrahydroquinoline to 3,4-dihydroquinoline by TiO2 [1] and (iii) reduction of nitrobenzene to aniline by Au/TiO2 [1], nitrophenol to aminophenol by N-TiO2 [2] and CdS [5], nitrotoluene to aminotoluene by N-TiO2 [2], nitroaniline to phenylenediamine by N-TiO2 [2] and CdS [5] and nitrobenzaldehyde to aminobenzaldehyde by N-TiO2 [2] have been reported. Diphenylamine (DPA) is used in post-harvest treatment of apple and pear [7] and photosensitized oxidation of DPA has been studied; cyanoanthracenes [8] and benzophenone [9] are some of the photosensitizers employed. The unsensitized photoconversion of DPA into N-phenyl-p-benzoquinonimine (PBQ) is slow [10]. Nanocrystalline ZnO is a unique functional oxide because of its dual semiconducting and piezoelectric properties. It has a wide range of applications in optics, optoelectronics, sensors, actuators, energy and biomedical sciences, and spintronics [11]. More importantly, it is a promising photocatalyst for environmental remediation [12] and is believed to be an alternative to TiO2 as both semiconductors possess approximately the same band gap. Furthermore, their conduction band (CB) energy levels and their valence band (VB) edges are not significantly different. In addition, there are reports that the photocatalytic activity of ZnO is better than that of TiO2 [13]. ZnO has also been employed as photocatalyst for organic transformation [1]. Hence it is of interest to study the photocatalytic transformation of DPA using nanocrystalline ZnO. It is possible to improve the photocatalytic activity by enhancing the lifetime of the charge carriers in semiconductor nanocrystals. This may be achieved through interparticle charge transfer (IPCT). If a mixture of two nanoparticulate semiconductors is employed as a photocatalyst for degradation of organic pollutants enhanced photocatalytic activity is observed [14, 15]. This has been explained invoking IPCT. However, such enhancement is not realized in the photocatalytic oxidation of iodide ion suggesting the IPCT to be slower than hole-transfer to iodide ion [16]. Hence it is of interest to study photocatalytic organic transformation using nanoparticulate semiconductor mixture as photocatalyst. Here we report enhanced photocatalytic organic transformation of DPA on mixing nanoparticulate TiO2 or V2O5 or CeO2 or ZnS with ZnO nanocrystals. Systematic study on ZnO-photocatalyzed transformation of DPA is necessary to obtain the kinetic law governing the reaction. The UV light (ca. 5%) present in the solar spectrum could photoexcite ZnO and hence the reaction has also been studied using natural sunlight.
Experimental
Materials and measurements
ZnO, TiO2, CeO2, V2O5 and ZnS (Merck) were used as supplied and their specific surface areas, obtained by BET method, are 12.2, 14.7, 11.0, 16.1 and 7.7 m2 g-1, respectively. The mean particle sizes (t) of ZnO, TiO2, CeO2, V2O5 and ZnS, obtained using the formula t = 6/ρS, where ρ is the material density and S is the specific surface area, are 87, 104, 76, 111 and 190 nm, respectively. The secondary particle size distributions occurring because of agglomeration in alcohol have been measured using particle sizer Horiba LA-910 or Malvern 3600E (focal length 100 mm, beam length 2.0 mm). The obtained powder X-ray diffractograms of ZnO, TiO2, CeO2 and ZnS show the crystalline structures as hexagonal wurtzite, tetragonal anatase, cubic fluorite and cubic zinc blende, respectively [17]. The UV-visible diffuse reflectance spectra of the materials were obtained using a Shimadzu UV-2600 spectrophotometer with ISR-2600 integrating sphere attachment. The Kubelka-Munk plots provide the band gaps of ZnO, TiO2, CeO2 and ZnS as 3.15, 3.18, 2.89 and 3.57 eV, respectively [17]. Potassium tris(oxalato)ferrate(III), K3[Fe(C2O4)3].3H2O, was prepared by standard method [18]. DPA, AR (Merck) was used as received. Commercial ethanol was purified by distillation with calcium oxide.
UV light-induced transformation
The UV light induced reaction on ZnO was carried out in a multilamp photoreactor fitted with eight 8 W mercury UV lamps (Sankyo Denki, Japan) of wavelength 365 nm, shielded by highly polished anodized aluminum reflector. Four cooling fans at the bottom of the reactor dissipate the generated heat. The reaction vessel was a borosilicate glass tube of 15-mm inner diameter and was placed at the center of the reactor. The UV light-induced reaction was also studied with a micro-photoreactor fixed with a 6 W 254 nm low-pressure mercury lamp and a 6 W 365 nm mercury lamp. Quartz and borosilicate glass tubes were employed as reaction vessels for 254 and 365 nm lamps, respectively. The light intensity (25.2 μeinstein L-1 s-1 unless otherwise mentioned) was determined by ferrioxalate actinometry. The volume of the reaction solution was always maintained as 25 mL in the multilamp photoreactor and 10 mL in the micro-photoreactor. Air was bubbled through the ethanolic solution of DPA (5 mmol L-1 unless otherwise stated) to keep the catalyst powder (1.0 g unless otherwise given) under suspension and at constant motion. The airflow rate (7.8 mL s-1 unless otherwise stated) was measured by soap bubble method. The UV-visible spectra were recorded using a Hitachi U-2001 UV-visible spectrophotometer. The solution was diluted 5-times to decrease the absorbance to the Beer-Lambert law limit. The PBQ formed was estimated from its absorbance at 450 nm.
Sunlight-induced transformation
The sunlight-induced reaction on ZnO was carried out under clear sky in summer (March-July) at 11.30 am-12.30 pm. The sunlight intensity (W m-2) was measured using a Global pyranometer (Industrial Meters, Bombay). The solar irradiance (einstein L-1 s-1) was also measured by ferrioxalate actinometry. The measured 440 W m-2 corresponds to 22 μeinstein L-1 s-1. Ethanolic solutions of DPA of required concentration (5.0 mmol L-1 unless otherwise stated) were prepared afresh for each set of experiments and taken in wide cylindrical glass vessels of uniform diameter. The entire bottom of the vessel (11.36 cm2 unless otherwise mentioned) was covered by ZnO powder (1.0 g). Air was bubbled (4.6 mL s-1 unless otherwise stated) with a micro-pump without disturbing the catalyst bed. The volume of DPA solution was 25 mL and the loss of solvent due to evaporation was compensated periodically. PBQ formed was estimated spectrophotometrically.
Results and Discussion
Photo-induced oxidative transformation on ZnO
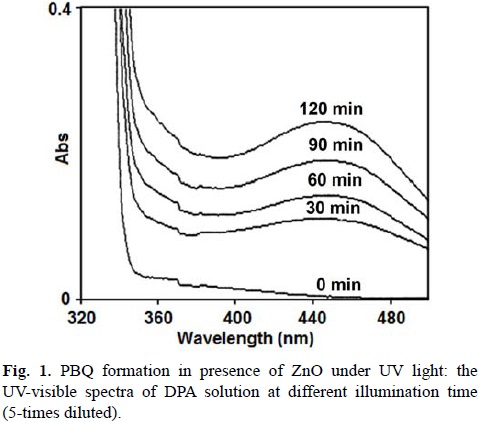
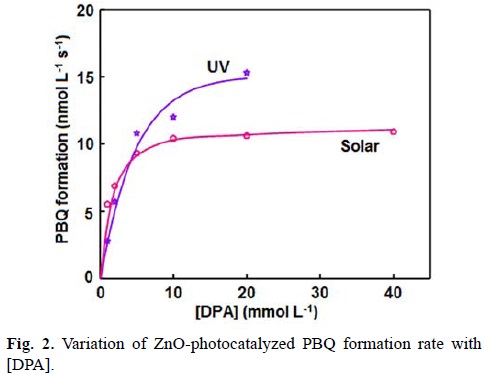
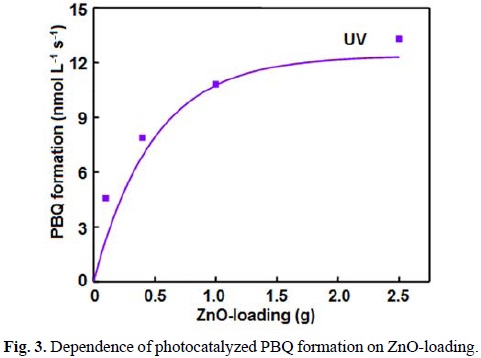
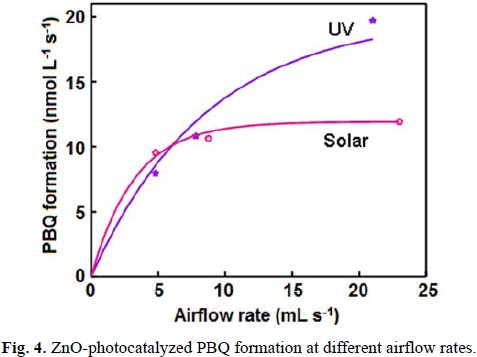
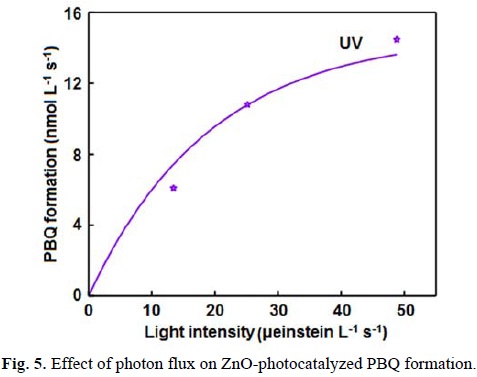
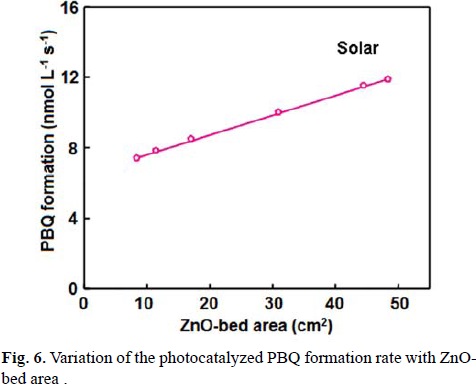
The UV light-induced oxidative transformation of DPA on ZnO surface in ethanol was performed in the presence of air using a multilamp photoreactor equipped with UV lamps of wavelength 365 nm. The UV-visible spectra of the DPA solution, recorded at different illumination time, reveal the formation of PBQ (λmax= 450 nm). Fig. 1 ([DPA]0 = 25 mmol L-1) displays the time spectra. The illuminated solution does not show electron paramagnetic resonance (EPR) signal indicating the absence of formation of diphenylnitroxide. Furthermore, thin layer chromatographic analysis reveals formation of a single product. The illuminated DPA solution was evaporated after the separation of ZnO and the solid was dissolved in chloroform to develop the chromatogram on a silica gel G-coated plate using benzene as eluent. The PBQ formed was estimated from the absorbance at 450 nm using its molar extinction coefficient [19, 20]. The linear increase of [PBQ] with illumination time (not shown) provides the initial rate of PBQ formation and the rates are reproducible to ±6%. The uncatalyzed photooxidation of DAP is slow [10] and the rate of PBQ formation on ZnO was obtained by measuring the rates of PBQ formation in the presence and absence of ZnO. The oxidative transformation of DPA into BPQ on ZnO surface takes place under natural sunlight also. The results obtained are similar to those with UV light. Measurement of the solar irradiance (W m-2) shows fluctuation of sunlight intensity during the experiment even under clear sky. Hence, the solar experiments at different reaction conditions were carried out in a set to maintain the quantity of sunlight incident on unit area the same. This makes possible to compare the solar results. A pair of solar experiments carried out simultaneously under identical reaction conditions yields results within ±6% and this is so on different days. The effect of operational parameters on the solar-induced oxidative transformation was investigated by carrying out the given set of experiments simultaneously and the results presented in each figure represent identical sunlight intensity. The rate of PBQ formation was obtained by illuminating the DPA solution on ZnO bed for 60 min. Fig. 2 shows the enhancement of PBQ formation on ZnO with [DPA]. The observed enhancement corresponds to Langmuir-Hinshelwood kinetics with respect to [DPA]. The rate of surface reaction with UV light increases with loading of ZnO in the DPA solution and the rate reach a limit at high ZnO-loading. The results are presented in Fig. 3. Measurement of the rate of PBQ formation on ZnO at different airflow rates shows enhancement of the surface reaction by oxygen and the rate dependence on the airflow rate conforms to the Langmuir-Hinshelwood kinetic law. Fig. 4 presents the results. The PBQ formation on ZnO was also determined without bubbling air but the solution was not deoxygenated. The dissolved oxygen itself brings in the light-induced surface reaction. However, the reaction is slow. PBQ formation on ZnO with UV light was examined at different light intensities (I). The reaction was performed with two, four and eight lamps and the angles sustained by the adjacent lamps are 180°, 90° and 45°, respectively. The dependence of the surface reaction rate on photon flux is shown in Fig. 5. PBQ is not formed in the dark. Study of the PBQ formation on ZnO with UV-A and UV-C light, employing a 6 W 365 nm mercury lamp (I=18.1 μeinstein L-1 s-1) and a 6 W 254 nm low-pressure mercury lamp (I=5.22 μeinstein L-1 s-1), separately in the micro-photoreactor under identical conditions shows that UV-C light is more efficient than UV-A light in inducing the organic transformation on ZnO. The PBQ formation with UV-A and UV-C light are 11.2 and 44.2 nmol L-1 s-1, respectively. The solar PBQ formation on ZnO increases linearly with the apparent area of ZnO-bed. Fig. 6 displays the results. ZnO retains its photocatalytic efficiency on usage. Reuse of ZnO shows sustainable light-induced PBQ formation. Singlet oxygen quencher azide ion (5 mmol L-1) does not inhibit the PBQ formation revealing the absence of involvement of singlet oxygen in the light-induced organic transformation on ZnO. This is in agreement with the literature; Fox and Chen [21] excluded the possibility of singlet oxygen in the TiO2-photocatalyzed olefin-to-carbonyl oxidative cleavage.
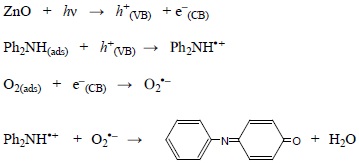
Mechanism
Band gap illumination of ZnO creates electron-hole pairs, electrons in the CB and holes in the VB. Recombination of the electron-hole pair is very fast, takes place in picosecond-time scale, and for an effective photocatalysis the reactants are to be adsorbed on the surface of ZnO. The hole is likely to pick up an electron from the surface adsorbed DPA molecule to form diphenylamine radical-cation (Ph2NH•+). The oxygen molecule adsorbed on the ZnO surface takes up the CB electron. The superoxide radical-anion formed probably reacts with diphenylamine radical-cation to afford PBQ.
Kinetic law
Kinetic analysis is possible with the results obtained using artificial UV light. The heterogeneous photocatalytic reaction taking place in a continuously stirred tank reactor (CSTR) conforms to the kinetic law [22]:
rate of PBQ formation on ZnO = kK1K2SIC[DPA]γ/(1+K1[DPA]) (1+K2γ)
where K1 and K2 are the adsorption coefficients of DPA and O2 on illuminated ZnO surface, k is the specific rate of oxidation of DPA on ZnO surface, γ is the airflow rate, S is the specific surface area of ZnO, C is the amount of ZnO suspended per litre and I is the photon flux. The obtained data fit to the Langmuir-Hinshelwood kinetic profile, drawn using a computer program [22] (Figs. 2 and 4). The linear double reciprocal plots of surface reaction rate versus [DPA] and airflow rate (not shown) also confirm the Langmuir-Hinshelwood kinetic law. The data-fit provides the adsorption coefficients K1 and K2 as 140 L mol-1 and 0.064 mL-1 s, respectively, and the specific reaction rate k as 28 μmol L m-2 einstein-1. However, the rate of PBQ formation on ZnO surface fails to increase linearly with the catalyst-loading. This is because of the high catalyst loading. At high catalyst loading, the surface area of the catalyst exposed to illumination does not correspond to the weight of the catalyst. The amount of ZnO used is beyond the critical amount corresponding to the volume of the reaction solution and the reaction vessel; the whole amount of ZnO is not exposed to light. The photoinduced transformation lacks linear dependence on illumination intensity; less than first power dependence of surface-photocatalysis rate on light intensity at high photon flux is well known [23].
Synergism by TiO2, V2O5, CeO2 and ZnS
In coupled semiconductors, also known as semiconductor composites, vectorial transfer of photoproduced charge carriers from one semiconductor to another is possible. This charge separation enhances the photocatalytic efficiency and examples for coupled semiconductors are many [24]. In coupled semiconductors, both the semiconductors coexist in the same particle and the charge separation occurs within the particle. But what we observe here is enhanced photoinduced transformation of DPA to PBQ on mixing nanoparticulate ZnS or TiO2 or CeO2 or V2O5 with ZnO nanoparticles. Fig. 7a presents the enhancement of photocatalytic formation of PBQ by ZnO mixed with ZnS or TiO2 or CeO2 or V2O5 illuminated by UV light - the two nanoparticulate semiconductors are in suspension and at constant motion. This observed enhanced photocatalytic transformation is likely due to interparticle charge transfer. Nanoparticles in suspension aggregate [25]. Fig. 7b displays the particle size distributions of ZnO, ZnS, TiO2, CeO2 and V2O5 nanoparticles in suspension.
Examination of Fig. 7b in conjunction with the determined particle sizes shows aggregation of the nanoparticles. As observed in individual nanoparticulate semiconductor suspension, aggregation is likely in nanoparticulate semiconductor mixtures under suspension and both the semiconductor nanoparticles are likely to be present in the aggregates. Charge transfer between ZnO and ZnS or TiO2 or CeO2 or V2O5 nanoparticles is likely to occur when both the semiconductors are under band gap-illumination and in contact with each other. Electron from CB of a semiconductor may move to another if the latter is of lower energy and so is the hole from VB. The CB electron of ZnO is less cathodic than those of ZnS [26] and CeO2 [27] and more cathodic than those of V2O5 and TiO2 [28]. This enables transfer of CB electron of ZnS and CeO2 to the CB of ZnO (in ZnO-ZnS and ZnO-CeO2 mixtures) and migration of CB electron of ZnO to the CB of TiO2 and V2O5 (in ZnO-TiO2 and ZnO-V2O5 mixtures). Similarly, the VB of ZnO is more anodic than those of ZnS and CeO2 and less anodic than those of TiO2 and V2O5. This favors hole-transfer from the VB of ZnO to those of ZnS and CeO2 (in ZnO-ZnS and ZnO-CeO2 mixtures) and hole-migration from VB of TiO2 and V2O5 to that of ZnO (in ZnO-TiO2 and ZnO-V2O5 mixtures). This interparticle charge transfer enhances the photocatalytic transformation. The energy difference between the CB electrons of the two semiconductors is the driving force for the interparticle electron separation and the free energy change is given by –ΔG = e(E(CBSC1)–E(CBSC2) [29]. In terms of redox chemistry, the CB and VB refer to the reduced and oxidized states in the semiconductor. In TiO2, CeO2,V2O5 and ZnO or ZnS the CB electrons refer to the reduced forms of Ti4+ (i.e., Ti3+), C4+ (i.e., Ce3+ V4+ (i.e.,V3+) and ZN2+ (i.e, Zn+), respectively. Similarly, the VB hole corresponds to the oxidized forms of the respective O2- (i.e., O-) or S2- (i.e., S-). The interparticle charge-transfer, the transfer of electron from the CB of ZnS or CeO2 to CB of ZnO refers to the electron jump from Zn+ of ZnS or Ce3+ to Zn2+ of ZnO. The electron jump from CB of ZnO to those of TiO2 and V2O5 corresponds to electron migration from Zn+ of ZnO to V5+ or Ti4+. The hole-transfer from the VB of ZnO to those of ZnS and CeO2 refers to the electron-jump from O2- of CeO2 or S2- of ZnS to O- of ZnO. Similarly, the hole-jump from the VB of TiO2 and V2O5 implies electron movement from O2- of ZnO to O- of TiO2 and V2O5. The possibility of cross-electron–hole combination, the transfer of electron from the CB of one semiconductor (SC1) to the VB of the other (SC2) is very remote; the very low population of the excited states renders the electron transfer between two excited states highly improbable. A possible reason for not observing the maximum photocatalytic transformation at 50% wt. composition of the semiconductor mixtures is the densities and particle sizes of the semiconductors and also the aggregation.
Conclusions
DPA on light-induced oxidative transformation on ZnO surface affords BPQ. The BPQ formation on ZnO increases with [DPA] and airflow rate and conforms to Langmuir-Hinshelwood kinetic law. The PBQ formation on ZnO is larger with UV-C light than with UV-A light. ZnO mixed with ZnS or TiO2 or CeO2 or V2O5 yields more PBQ than by the individual semiconductor and this is likely to be due to interparticle charge transfer.
Acknowledgement
Prof. C. Karunakaran is thankful to the Council of Scientific and Industrial Research (CSIR), New Delhi for the Emeritus Scientist Scheme [21(0887)/12/EMR-II].
References
1. Lang, X.; Chen, X.; Zhao, J. Chem. Soc. Rev. 2014, 43, 473-486. [ Links ]
2. Palmisano, G.; Garcia-Lopez, E.; Marci, G.; Loddo, V.; Yurdakal, S.; Augugliaro, V.; Palmisano, L. Chem. Commun. 2010, 46, 7074-7089. [ Links ]
3. Shiraishi, Y.; Hirai, T. J. Photochem. Photobiol. C. 2008, 9, 157-170. [ Links ]
4. Feng, W.; Wu, G.; Li, L.; Guan, N. Green Chem. 2011, 13, 3265-3272. [ Links ]
5. Yang, M.-Q.; Xu, Y.-J. Phys. Chem. Chem. Phys. 2013, 15, 19102-19118. [ Links ]
6. Zhang, N.; Liu, S.; Fu, X.; Xu, Y.-J. J. Mater. Chem. 2012, 22, 5042-5052. [ Links ]
7. Zanella, A. Postharvest Biol. Technol. 2003, 27, 69-78. [ Links ]
8. Chang, Y. C.; Chang, P. W.; Wang, C. M. J. Phys. Chem. B. 2003, 107, 1628-1633. [ Links ]
9. Lin, T. S.; Retsky, J. J. Phys. Chem. 1986, 90, 2687-2689. [ Links ]
10. Karunakaran, C.; Karuthapandian, S. Sol. Energy Mater. Sol. Cells. 2006, 90, 1928-1935. [ Links ]
11. Wang, Z. L. Mater. Sci. Eng. R. 2009, 64, 33-71. [ Links ]
12. Wang, X.; Zhang, Q.; Wan, Q.; Dai, G.; Zhou, C.; Zou, B. J. Phys. Chem. C. 2011, 115, 2769-2775. [ Links ]
13. Li, Y.; Xie, W.; Hu, X.; Shen, G.; Zhou, X.; Xiang, Y.; Zhao, X.; Fang, P. Langmuir. 2010, 26, 591-597. [ Links ]
14. Karunakaran, C.; Dhanalakshmi, R.; Gomathisankar, P. Int. J. Chem. Kinet. 2009, 41, 716-726. [ Links ]
15. Karunakaran, C.; Dhanalakshmi, R.; Gomathisankar, P.; Manikandan, G. J. Hazard. Mater. 2010, 176, 799-806. [ Links ]
16. Karunakaran, C.; Anilkumar, P.; Vinayagamoorthy, P. Spectrochim. Acta A. 2012, 98, 460-465. [ Links ]
17. Karunakaran, C.; Gomathisankar, P.; Manikandan, G. Indian J. Chem. Technol. 2011, 18, 169-176. [ Links ]
18. Adams, D. M.; Raynor, J. B. Advanced Practical Inorganic Chemistry, John Wiley, New York, 1965, 54. [ Links ]
19. Puri, S.; Bansal, W. R.; Sidhu, K. S. Indian J. Chem. 1973, 11, 828. [ Links ]
20. Bansal, W. R.; Ram, N.; Sidhu, K. S. Indian J. Chem. B. 1976, 14, 123-126. [ Links ]
21. Fox, M. A.; Chen, C. C. J. Am. Chem. Soc. 1981, 103, 6757-6759. [ Links ]
22. Karunakaran, C.; Senthilvelan, S.; Karuthapandian, S. J. Photochem. Photobiol. A. 2005, 172, 207-213. [ Links ]
23. Vincze, L.; Kemp, T. J. J. Photochem. Photobiol. A. 1995, 87, 257-260. [ Links ]
24. Karunakaran, C.; SakthiRaadha, S.; Gomathisankar, P.; Vinayagamoorthy, P. RSC Adv. 2013, 3, 16728-16738. [ Links ]
25. Li, M.; Noriega-Trevino, M. E.; Nino-Martinez, N.; Marambio-Jones, C.; Wang, J.; Damoiseause, R.; Ruiz, F.; Hock, E. M. V. Environ. Sci. Technol. 2011, 45, 8989-8995. [ Links ]
26. Fan, W.; Zhang, Q.; Wang, Y. Phys. Chem. Chem. Phys. 2013, 15, 2632-2649. [ Links ]
27. Tian, J.; Sang, Y.; Zhao, Z.; Zhou, W.; Wang, D.; Kang, X.; Liu, H.; Wang, J.; Chen, S.; Cai, H.; Huang, H. Small. 2013, 9, 3864-3872. [ Links ]
28. Lei, Y.; Zhao, G.; Liu, M.; Zhang, Z.; Tong, X.; Cao, T. J. Phys. Chem. C. 2009, 113, 19067-19076. [ Links ]
29. Katoh, R.; Furube, A.; Yoshihara, T.; Hara, K.; Fujihashi, G.; Takano, S.; Murata, S.; Arakawa, H.; Tachiya, M. J. Phys. Chem. B. 2004, 108, 4818-4822. [ Links ]

