










Servicios Personalizados
Revista
Articulo
 Inglés (pdf)
Inglés (pdf)
 Artículo en XML
Artículo en XML Referencias del artículo
Referencias del artículo
 Enviar artículo por email
Enviar artículo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO -
 Accesos
Accesos
Links relacionados
-
 Similares en
SciELO
Similares en
SciELO
Compartir

 Permalink
PermalinkJournal of the Mexican Chemical Society
versión impresa ISSN 1870-249X
J. Mex. Chem. Soc vol.59 no.1 Ciudad de México ene./mar. 2015
Article
Hydrogen Release from NH3 in the Presence of BN Graphene: DFT Studies
Ali Ahmadi Peyghan,1 Hamed Soleymanabadi,2 and Zargham Bagheri3*
1 Young Researchers and Elite club, Central Tehran Branch, Islamic Azad University, Tehran, Iran.
2 Department of Chemistry, College of Science, Central Tehran Branch, Islamic Azad University, Tehran, Iran.
3 Department of Physics, College of Science, Islamshahr Branch, Islamic Azad University, Islamshahr, Iran. Bagherizargham@gmail.com
Received April 24th, 2014
Accepted December 11th, 2014
Abstract
Using density functional theory, we investigated the interaction of an NH3 molecule with a pristine and antisite defected BN sheet (g-BN) in terms of energetic, geometric, and electronic properties. The adsorption energy of NH3 on defected g-BN was calculated to be in the range of -0.70 to -2.46 eV, which is considerably more negative than that on the pristine sheet. It was found that the adsorption of NH3 adsorption on the defected sheet may cause the release of an H2 molecule. The electronic properties of the defected BN sheet were significantly changed after the adsorption process so that its HOMO/LUMO energy gap was changed from 3.31 to 3.60-4.97 eV. Moreover, the Fermi level of the defected sheet shifts to higher energies after the interaction, which results in reduced potential barrier of the electron emission for the sheet surface, enhancing the field emission because of the decreased work function.
Key words: Boron Nitride Nanosheet, Graphene-like, DFT, B3LYP, Ammonia.
Resumen
Usando la teoría de funcionales de la densidad investigamos la interacción de una molécula de NH3 con sábanas de grafeno prístinas y con defectos anti-sitio BN (g-BN) en términos energéticos, geométricos y de propiedades electrónicas. La energía de absorción calculada de NH3 en g-BN está en el rango de -0.70 a -2.46 eV, lo cual es considerablemente más negativo que en las sábanas prístinas. Se encontró que la absorción de NH3 en las sábanas con defectos puede causar la liberación de una molécula de H2. Las propiedades electrónicas de las sábanas con defectos BN muestran que hay carga significativa después del proceso de absorción, de modo que la diferencia de energía HOMO/LUMO varía de 3.31 a 3.60-4.97 eV. Por otra parte, el nivel de Fermi de las sábanas con defecto se mueve a energías de interacción más altas lo que resulta en barreras de energía potencial reducidas para la emisión de electrones desde la superficie de la sábana, incrementado el campo de emisión debido a la disminución de la función trabajo.
Palabras clave: Nano-sábanas con Boro y Nitrógeno, tipo grafeno, TDF, B3LYP, amonio.
Introduction
Depletion of fossil fuels as well as environmental concerns has led to widespread research for finding new and renewable energy sources. Hydrogen (H2) has attracted great interest because of its high chemical energy content (at least three times larger than that of other chemical fuels), abundance, and non-polluting nature [1, 2]. Hydrogen economy offers an alternative to our dependence on fossil fuels and provides an environmentally clean source of energy. However, the application of hydrogen economy for practical applications requires efficient H2 storage with high gravimetric and volumetric density [3]. Therefore, much research has been focused on hydrogen production from hydrogen storage materials such as CaH2, LiH, NaBH4, MgH2, LiAlH4, KBH4, NaAlH4, and NH3 with superior catalyst systems [4, 5].
Ammonia contains 17.8 wt% hydrogen and stores 30% more energy by liquid volume than liquid hydrogen. Ammonia is the second largest synthetic commodity product of the world's chemical industry and the infrastructure for ammonia transportation, distribution, storage, and utilization is well-established [6]. It is also very important to note that NH3 molecules are carbon free; there is no need for the purification of hydrogen produced by NH3 decomposition because its by-product most often is N2 that is benign. Ammonia can be used as an on-board carrier of hydrogen, where the latter can be released by NH3 decomposition.
Ever since the discovery of carbon nanotubes (CNTs) [7], there have been significant research efforts to synthesize nanometer scale tubular forms of various materials and to study their properties [8-13]. A sheet of carbon atoms arranged in a two-dimensional hexagonal lattice is known as graphene. Graphene sheets can be transformed into various dimensional carbon materials by self-assembly and are thus the (2D) building blocks of many carbon materials [14-16]. Nevertheless, graphene usually suffers from various types of defect in atomic structures during its growth. A large number of experimental and theoretical studies have demonstrated that the existence of defects, such as Stone-Wales defects, vacancies, antisite, and pentagonal-octagon in graphene and nanotubes can drastically change the properties of these nanostructures [17-23]. Similar to graphene, graphene-like BN (g-BN) as a wide gap semiconductor, has been successfully fabricated by controlled energetic electron irradiation through a layer-by-layer sputtering process [24]. The g-BN has aroused extensive research interest because of its many intriguing properties such as high chemical stabilities, excellent mechanical properties, and high thermal conductivities [25-28]. Compared to their carbon counterparts, g-BN nanomaterials have much larger band gap, hence low electrical conductance. The surface of g-BN nanomaterials is intrinsically inert because of the large ionicity of the BN bond, making the chemical modification and practical processing of these materials difficult.
In the present work, the interaction of NH3 with g-BN will be theoretically investigated based on analyses of structure, energies, electronic properties, etc. To this end, we have tried to find the answers of the following questions: (1) potential utility of g-BN as a H2 generator from NH3; (2) if not, what strategy can be applied to improve the reactivity of the g-BN toward NH3.
Computational Methods
A g-BN consisting of 36 boron and 36 nitrogen atoms was considered, whose end atoms were saturated with hydrogen atoms to avoid the boundary effects. The full geometry optimizations and property calculations on the pristine and antisite defected g-BN in the presence and absence of a NH3 molecule were performed using three-parameter hybrid generalized gradient approximation with the B3LYP functional and the 6-31G basis set including the d-polarization function (denoted as 6-31G (d)) as implemented in the GAMESS suite of program [29]. GaussSum program [30] was used to obtain DOS results. In order to plot DOS plots, the orbital energies were set to be -10 to 4 eV and the full width at half maximum was 0.3 Å. The B3LYP density functional has been previously shown to reproduce experimental proprieties and has been commonly used for nanostructures [31-34]. It has also been demonstrated that the B3LYP provides an efficient and robust basis for calculations of III-V semiconductors by Tomić et al. [35], capable of reliably predicting both the ground state energies and the electronic structure. The adsorption energy (Ead) of an NH3 molecule on the g-BN is obtained using the following equation:
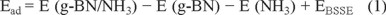
where E (g-BN/NH3), E (NH3), and E (g-BN) are referred to the energies of the g-BN/NH3 complex, isolated NH3 molecule, and the pristine or defected g-BN, respectively. EBSSE is the energy of the basis set superposition error. The negative value of Ead indicates the exothermic character of the adsorption. The canonical assumption for Fermi level (EFL) is that in a molecule (at T = 0 K) it approximately is average of the HOMO-LUMO energy gap (Eg).
Results and discussion
NH3 adsorption on the pristine g-BN
The optimized structure of the pristine g-BN is shown in Fig. 1, in which the lengths of B-N bonds are in the range of 1.42-1.48 Å, in good accordance with previous experimental and theoretical results [36]. Calculated density of states (DOS) plot shows that the g-BN is a semiconductor with the Eg of 5.93 eV (Fig. 1). In order to find minimum adsorption configurations, the NH3 molecule was initially placed at different positions above a g-BN, on top of either boron or nitrogen atoms from its H or N atoms. After relax geometry optimization of initial guesses, only one stable NH3 sheet was predicted (Fig. 2). Other initial configurations have reoriented to this stable structure during the relax optimization. In this configuration, the N atom of NH3 forms a chemical bond with a B atom of the sheet (Fig. 2) with Ead of -0.40 eV (Table 1). Our NBO calculations show that the hybridization of the B atom in the pure sheet and complex form is about sp2.04 and sp2.11, respectively. The adsorption of NH3 on the B site of g-BN may be explained by the fact that the B site acts as a Lewis acid for the base of NH3. Calculated DOS plots (Fig. 2) show that the NH3 adsorption through this configuration has no sensible effects on the electronic properties of the sheet so that the Eg of the sheet has slightly decreased from 5.93 to 5.65 eV.
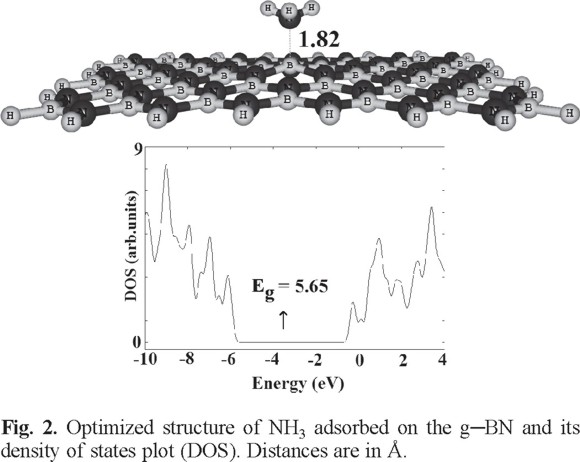

NH3 adsorption on the antisite defected g-BN
Next, we have studied the interaction of a defected g-BN with an NH3 molecule. Among various native defects, we have focused on boron antisite (BN), in which a boron atom sits at the original nitrogen site, which is surrounded by three boron atoms, as shown in Fig. 3. The calculated bond length for the B-B bonds is 1.60 Å, which is much longer than the corresponding B-N bonds. Although the geometric structure of the g-BN is slightly distorted by substituting the N atom with the impurity of B (antisite defect), the optimized geometry for this defected system is perfectly planar. As a result, the B-B-B angle in the defected g-BN is 120°, which is similar to B-N-B in the pristine sheet (almost 120°).
Subsequently, we have explored NH3 adsorption on the defected site by locating the molecule above the impurity atom with different initial orientations including H or N atom of the molecule which is close to the BN atom. We have also put the molecule on a B-B bond so that the H and N atoms are located on the top of the B atoms. However, after relax optimization of the initial structures, five stable configurations were obtained which have been shown in Figs. 4, and 5. We have divided the interactions, according to these minima structures, into two types: (I) chemisorption, in which the Ead value is rather large in comparison with the pristine g-BN, and an insignificant change occurs in the geometrical parameters (configurations A and B, Fig. 4); and (II) dissociation, a very strong interaction that largely deforms the structure of the sheet and the molecule with bond cleavage and formation (configurations C and D, Fig. 4). Also, in one of the dissociation configurations, NH3 molecule strongly interacts with the defected g-BN and one H2 molecule was released (configuration E, Fig. 5). The Ead, charge transfer, and Eg for all configurations have been summarized in Table 2.
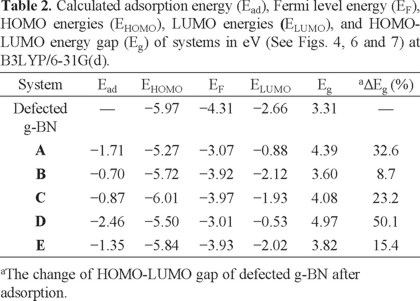
Configuration A stands for the covalent bonding between the nitrogen atom of the NH3 and the impure BN atom of the sheet with the distance of 1.65 Å, and its corresponding calculated Ead value is about -1.71 eV (Fig. 4A). The large Ead of NH3 on defected g-BN in this structure and the small distance between them reveal the chemical nature of the interaction. The BN atom bonded to NH3 molecule is lifted up, which, as the NBO analysis suggests, can be attributed to the change of their hybridization from sp2 to nearly sp3. In addition, the length of BN-B is increased from 1.60 Å to 1.67 Å after NH3 adsorption. As shown in Fig. 4B, configuration B is the second most stable one for NH3 chemisorption on defected g-BN system, in which the N atom of the molecule is close to a B atom of the sheet (which is the neighbor of BN), with equilibrium distance of 1.70 Å and corresponding Ead of -0.70 eV. Favorability of NH3 adsorption on defected g-BN than on the pristine one can be explained by the fact that in defected g-BN the LUMO is mainly located on the B atoms of the defected site (Fig. 6). As a result, the HOMO of NH3, located on the N atom, donates electrons preferentially to the LUMO centered on the B sites.
The calculated DOS of the defected g-BN is shown in Fig. 3, indicating that its Eg value is reduced to 3.31 eV, compared to the pristine g-BN. However, the defect forms an acceptor-like level, revealing that introducing a BN antisite defect will create a p-type semiconductive material with increased conductivity. Interestingly, the DOS plot of configuration A shows a considerable change near the Fermi level, indicating that the electronic properties of the defected g-BN are very sensitive to the NH3 adsorption. Valence level in this configuration is approximately similar to that of the defected sheet, while the conduction level significantly shifts downwards. As can be seen in Fig. 4A, the Eg value of the defected g-BN is dramatically increased from 3.31 eV to 4.39 eV (by about 34.3% change) in the adsorbed form, which would result in a change in the electrical conductivity of the defected sheet.
Subsequently, we have explored NH3 dissociation by locating the molecule above the defected g-BN. The double-acceptor B-B antisite can interact with an NH3 molecule strongly enough to break one of the N-H bonds. This type of interaction is analogous with the dihydrogen interaction in boron-doped carbon nanostructures [37], where a closed-shell H2 is attracted by a single acceptor state of the carbon-substituted boron. We have identified two distinct configurations for the NH3 dissociation, as shown in Fig. 4, namely C and D. In these configurations, N-H bond of the molecule is located on the top of the active BN-B bond and NH3 dissociation occurs. The main difference between these configurations is the orientation of -NH2 and -H fragments of the NH3. In both configurations, two new B-H and B-NH2 bonds are formed with the lengths of 1.21 Å (1.21 Å) and 1.47 Å (1.49 Å), respectively. As a result, the Ead of NH3 on the sheet in configuration D (-2.46 eV) is remarkably more than that of the configuration C (-0.87 eV).
Surface modification is a commonly used method to tailor the physical and chemical properties of BN nanostructures. However, low chemical reactivity of the surface of g-BN means that the chemical modification of the structure is much more difficult than that of the graphene. Calculated DOS plots for configurations C and D have been shown in Fig. 5, indicating that the NH3/defected g-BN complexes of configurations C and D attain E g values of 4.08 and 4.97 eV, respectively, while Eg of the defected g-BN is about 3.31 eV (Fig. 3). It can be therefore concluded that the modification of the defected g-BN by NH3 molecule obviously changes the electrical properties of the sheet. Theoretical investigations have shown that introducing adsorbates on graphene surface might effectively modify the field emission properties of the material [38], which is essential to estimate the potential for designing efficient field emission displays. As shown in Table 2, not only the Eg of the defected g-BN has been changed, but also the EFL has increased from -4.31 to -3.07 eV in configuration A. This phenomenon leads to a decrement in the work function which is important in field emission applications. The work (Φ) function can be found by :

where - e is the charge of an electron, V is the electrostatic potential in the vacuum nearby the surface, and EF is the energy of Fermi level. The work function is thus defined as the work required to remove an electron from the material to a state at rest in the vacuum nearby the surface. The decrement in the work function indicates that the field emission properties of the defected g-BN are facilitated after the adsorption of the NH3 molecules.
Fig. 5 shows the chemical functionalization of the defected g-BN by NH3. In this configuration, -NH fragment has diffused into the defected site from its N atom, so that two new BN-N and B-N bonds are formed with the bond lengths of 1.42 and 1.46 Å, respectively. Interestingly, an H2 molecule in this configuration has escaped from the sheet of defected g-BN. In this configuration (E), the Ead value is about -1.35 eV, somewhat weaker than that of C and D configurations. We think that the more positive value of Ead in this configuration can be attributed to the large deformation of NH3 and also the sheet after the adsorption process. The results indicate that our proposed system may be a promising material for hydrogen storage. We also calculated the barrier energy for this process. Transition state structure is shown in Fig. 7, which has been optimized using the Synchronous Transit-Guided Quasi-Newton Method. The calculation shows that releasing an H2 molecule needs the reaction process to overcome an energy barrier of 0.65 eV, which is relatively large. Thus, we think that these reactants may need to be heated to higher than room temperature to surmount this barrier energy. Also, our calculation shows that energy of 4.68 eV is needed to remove the -NH from the g-BN which is very high .We believe that the large energy is because of high instability of free -NH group. In our opinion, removal of this radical might be facilitated by using an auxiliary chemical group which needs more research.
For further investigation of the electronic properties of this new material, we have removed NH3 molecule from the surface of g-BN and full relax optimization was performed. As shown in Fig. 5, the DOS of the NH-functionalized g-BN has a change near the conduction level compared to that of the defected g-BN, so that the LUMO energy shifts from -2.66 eV to -2.02 eV. In this structure, the Eg of the defected g-BN is increased to 3.82 eV (with 15.4% change) in the functionalized form. Our results have already provided an important insight into the difference in behavior of the pristine and antisite defected g-BN towards NH3 adsorption and dissociation. Thus, we believe that introducing an antisite defect in g-BN can be a good strategy for improving the H2 production by NH3, which cannot be released by the pristine g-BN.
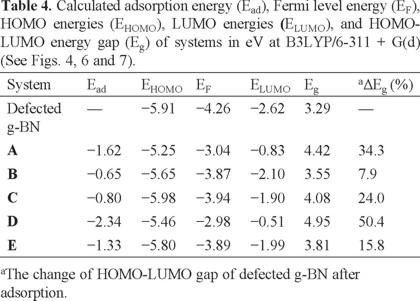
Finally, we have repeated our calculations with a larger basis set or a larger sheet. To this end, a g-BN consisting of 72 boron and 72 nitrogen atoms was considered. Our results show that Eg of systems slightly decreased in the range of 0.06 and 0.11 eV. The change of Ead is in the range of 0.01-0.04 eV which is negligible. Once again calculation on the former sheet was repeated by using B3LYP with 6-311 + G(d). The results (Tables 3 and 4) indicates that absolute values of calculated parameter slightly changed in comparison to 6-31G(d) basis set (Tables 1 and 2).

Conclusion
The geometric structures and electronic properties of the pristine and antisite defected g-BN in the presence and absence of an adsorbed NH3 molecule have been explored using density functional theory. It was found that the NH3 molecule weakly interacts with the pristine g-BN, but it presents much higher reactivity towards the antisite defected g-BN. Ead of NH3 on defected g-BN was calculated to be in the range of -0.70 to -2.46 eV. It suggests that the antisite defected g-BN can be a promising surface for dissociation of NH3 for chemical hydrogen storage. Also, the work function of the defected sheet is reduced after the interaction, resulting in reduced potential barrier of the electron emission for the sheet surface, hence enhancing the field emission.
References
1. Ni, M.; Leung, M.K.H.; Sumathy, K.; Leung, D.Y.C. Int. J. Hydrogen Energy 2006, 31, 1401-1412. [ Links ]
2. Baei, M.T.; Peyghan, A.A.; Bagheri, Z. C.R. Chimie 2013, 16, 122-128. [ Links ]
3. Goltsov, V.A.; Veziroglu, T.N.; Goltsov, L.F. Int J Hydrogen Energy 2006, 31, 153-159. [ Links ]
4. Chen, P.; Xiong, Z.; Luo, J.; Lin, J.; Tan, K.L. Nature 2002, 420, 302-304. [ Links ]
5. Amendola, S.C.; Sharp-Goldman, S.L.; Janjua, M.S.; Kelly, M.T.; Petillo, P.J.; Binder, M. J. Power Sources 2000, 85, 186189. [ Links ]
6. Raissi, A.T.; Danz, R. Analysis of hydrogen production using ammonia and ammonia-borane complex for fuel cell applications. Hydrogen, Fuel Cells, and Infrastructure Technology Program, FY, Progress Report, DOE, 2002. [ Links ]
7. Iijima, S. (1991) Nature 354, 56-58. [ Links ]
8. Peyghan, A.A.; Omidvar, A.; Hadipour, N.L.; Bagheri Z.; Kamfiroozi, M. Physica E 2012, 44, 1357-1360. [ Links ]
9. Ahmadi, A.; Hadipour, N.L.; Kamfiroozi, M.; Bagheri, Z. Sens. Actuat B-Chem. 2012, 161, 1025-1029. [ Links ]
10. Rogel-Hernandez, E.; Alonso-Nonez, G.; Camarena, J.P.; Espinoza-Gomez, G.A.; Paraguay-Delgado, P.; Somanthan, R. J. Mex. Chem. Soc. 2011, 55, 7-10. [ Links ]
11. Alonso, J.A.; Cabria, I.; Lopez, M.J. J. Mex. Chem. Soc. 2012, 56, 261-269. [ Links ]
12. Beheshtian, J.; Kamfiroozi, M.; Bagheri, Z.; Ahmadi, A. Physica E 2011, 44, 546-549. [ Links ]
13. Beheshtian, J.; Peyghan, A. A.; Bagheri, Z. J. mol. model. 2012, 19, 391-396. [ Links ]
14. Novoselov, K.S.; Geim, A.K.; Morozov, S.V.; Jiang, D.; Zhang, Y.; Dubonos, S.V.; Grigorieva, I.V.; Firsov, A.A. Science 2004, 306, 666-669. [ Links ]
15. Novoselov, K.S.; Geim, A.K.; Morozov, S.V.; Jiang, D.; Katsnelson, M.I.; Grigorieva, I.V.; Dubono, S.V.; Firsov, A.A. Nature 2005, 438, 197-200. [ Links ]
16. Wang, Z.; Gao, Y.; Xia, J.; Zhang, F.; Xia, Y.; Li, Y. C.R. Chimie 2012, 15, 708-713. [ Links ]
17. Peyghan, A. A.; Hadipour, N.; Bagheri, Z., J. Phys. Chem. C 2013, 117, 2427-2432. [ Links ]
18. Beheshtian, J.; Peyghan, A. A.; Bagheri, Z., Struct. Chem. 2013, 24, 1565-1570. [ Links ]
19. Peyghan, A. A.; Moradi, M., Thin Solid Films 2014, 552, 111-115. [ Links ]
21. Lehtinen, P.O.; Foster, A.S.; Ayuela, A.; Krasheninnikov, A.; Nordlund, K.; Nieminen, R.M. Phys. Rev. Lett. 2003, 91, 017202-017206. [ Links ]
22. Choi, H.; Park, Y.C.; Kim, Y. H.; Lee, Y.S. J. Am. Chem. Soc. 2010, 133, 2084-2087. [ Links ]
23. Peyghan, A. A.; Moradi, M., J. Mol. Model. 2014, 20, 1-7. [ Links ]
24. Nag, A.; Raidongia, K.; Hembram, K.P.S.S.; Datta, R.; Wanghmare, U.V.; Rao, C.N.R. ACS Nano 2010, 4, 1539-1544. [ Links ]
25. Beheshtian, J.; Soleymanabadi, H.; Peyghan, A.A.; Bagheri, Z. Appl. Surf. Sci. 2012, 268, 436-441. [ Links ]
26. Preobrajenski, A.B.; Nesterov, M.A.; Ng, M.L.; Vinogradov, A.S.; Mårtensson, N. Chem. Phys. Lett. 2007, 446, 119-123. [ Links ]
27. Beheshtian, J.; Soleymanabadi, H.; Peyghan, A. A.; Bagheri, Z., Applied Surface Science 2012, 268, 436-441. [ Links ]
28. Peyghan, A. A.; Noei, M.; Yourdkhani, S., Superlattices Microstruct. 2013, 59, 115-122. [ Links ]
29. Schmidt, M.W.; Baldridge, K.K.; Boatz, J.A.; Elbert, S.T.; Gordon, M.S.; Jensen, J.H.; Koseki, S.; Matsunaga, N.; Nguyen, K.A.; Su, S.; Windus, T.L.; Dupuis, M.; Montgomery, J.A. J. Comput. Chem. 1993, 14, 1347-1363. [ Links ]
30. O'Boyle N. M.; Tenderholt A.L.; Langner K.M., J. Comput. Chem. 2008, 29, 839-845. [ Links ]
31. Soltani, A.; Taghartapeh, M.R.; Tazikeh Lemeski, E.; Abroudi, M.; Mighani, H. Superlattic. Microstruct. 2013, 58, 178-190. [ Links ]
32. Pan, S.; Banerjee, S.; Chattaraj, P.K. J. Mex. Chem. Soc. 2012, 56, 229-240. [ Links ]
33. Owens, F.J. Matter. Lett. 2007, 61, 1997-1999. [ Links ]
34. Peyghan, A.A.; Noei, M. J. Mex. Chem. Soc. 2014, 58, 46-51. [ Links ]
35. Tomić S.; Montanari B.; Harrison N. M. Physica E 2008, 40, 2125-2127. [ Links ]
36. Golberg D.; Bando Y.; Huang Y.; Terao T.; Mitome M.; Tang C.; Zhi C. Acs Nano 2010, 4, 2979-2993. [ Links ]
37. Kim Y. H.; Zhao Y.; Williamson A.; Heben M. J.; Zhang S. B. Phys. Rev. Lett. 2006, 96, 016102-016106. [ Links ]
38. Ye D.; Moussa S.; Ferguson J. D.; Baski A. A.; El-Shall M. S. Nano Lett. 2012, 12, 1265-1268. [ Links ]

