










Servicios Personalizados
Revista
Articulo
 Inglés (pdf)
Inglés (pdf)
 Artículo en XML
Artículo en XML Referencias del artículo
Referencias del artículo
 Enviar artículo por email
Enviar artículo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO -
 Accesos
Accesos
Links relacionados
-
 Similares en
SciELO
Similares en
SciELO
Compartir

 Permalink
PermalinkJournal of the Mexican Chemical Society
versión impresa ISSN 1870-249X
J. Mex. Chem. Soc vol.59 no.1 Ciudad de México ene./mar. 2015
Article
Theoretical Study of the Formation of Complexes Between CO2 and Nitrogen Heterocycles
Elizabeth Hernández-Marín, and Ana Adela Lemus-Santana*
Centro de Investigación en Ciencia Aplicada y Tecnología Avanzada, Unidad Legaria, Instituto Politécnico Nacional, México D.F. ehdzmarin@gmail.com, alemuss@ipn.mx
Received July 10th, 2014
Accepted November 4th, 2014
Abstract
A density functional theory study was performed to analyze the formation of complexes between CO2 and different nitrogen heterocycles such as imidazole, 2-methylimidazole, benzimidazole, and pyrazine. Two orientations of CO2 were considered: in-plane and top-on with respect to the plane of the heterocyclic ring. The in-plane complexes are more stable than their top-on counterparts, most likely due to electrostatic and Lewis acid-base interactions. The strength of the intermolecular interactions in the top-on complexes can be related to a combination of dispersion, weak electrostatic, dipole-quadrupole and quadrupole-quadrupole interactions, and to some extent to the interactions where some charge transfer from the ring to CO2 is involved. With respect to a potential use as CO2 scrubbers, imidazole and its derivatives appear to be better than pyrazine.
Keywords: CO2 scrubbers, DFT, Dispersion Corrections, Imidazole Derivatives, Pyrazine.
Resumen
Se presenta un estudio basado en la teoría de funcionales de la densidad que analiza la formación de complejos entre CO2 y diferentes heterociclos de nitrógeno tales como imidazol, 2-metilimidazol, benzimidazol y pirazina. Fueron consideradas dos orientaciones del CO2 con respecto al plano anillo heterocíclico: en-el-plano y sobre-el-plano. Los complejos en-el-plano son más estables que sus contrapartes sobre-elplano, debido a la influencia de interacciones electrostática y de tipo ácido-base de Lewis. Las fuerzas de las interacciones intermoleculares en los complejos sobre-el-plano se pueden relacionar con una combinación entre fuerzas de dispersión, interacciones electrostáticas débiles y otras como las de tipo dipolo-cuadrupolo y cuadrupolo-cuadrupolo y hasta cierto punto con interacciones donde algo de carga se transfiere del anillo a la molécula de CO2. Con respecto a un uso potencial para la captura de CO2, el imidazol y sus derivados parecen ser mejores que la pirazina.
Palabras clave: Captura de CO2, TFD, correcciones con el término de dispersión, derivados de imidazol, pirazina.
Introduction
According to the United States Environmental Protection Agency (EPA), carbon dioxide (CO2) capture and sequestration could play an important role in reducing greenhouse gas emissions [1]. Carbon capture and sequestration (CCS) technologies involve, among others, the compression of CO2, its transportation and subsequent underground injection for permanent storage. Some technologies employed to remove CO2 from air include the use of NaOH and KOH [2], while the most developed technologies make use of aqueous solutions of alkalo-amines [3], where the regeneration of the amine requires a high energy input [2-4].
Current alternative technologies for CO2 capture include the use of porous inorganic membranes (PIMs) [5], metal organic frameworks (MOFs) [6,7] and zeolitic imidazole frameworks (ZIFs) [8] type materials.
Host lattices of Hofmann clathrate are known by the selectivity presented toward different molecules, as well as for the stereochemical preferences of enclathrated molecules, for example in Hofmann clathrates. This feature impelled some theoretical studies on the nature of the host-guest interactions [9-11]. When aromatic dinitrogen ligands (pillar ligands) were used, galleries became separated channels with tunable sizes and functionality [12, 13]. Thus, forty coordination polymers referred to as pillared cyanonickelates (PICNICs) [14] have been screened as CO2 sorbents. As a different approach to implement functionalization to the host lattice, we focused on the direct coordination of organic molecules to a T metal in the 2D grid. In this way, some new materials with formula TL2[Ni(CN)4] (T = Ni2 + , Co2 + or Mn2 + ; L = Imidazole, Benzimidazole, 2-methylimidazole, Pyrazine, etc.) have been recently synthesized and characterized in our group [15-17]. In those solids it was found that the L ligands are located in between quasi-parallel T[Ni(CN)4] layers. The remarkable feature in these families is the combination of layers flexibility with the incorporation of heteroatoms in organic rings, which creates very different chemical ambient as a whole. As a first step to study these families as carbon dioxide sorbents we consider worthwhile to explore, the feasibility of the formation of adducts between the above-mentioned heterocycles and CO2 from a theoretical point of view. Previous theoretical works have already studied complexes with aminoacids [18, 19], amines [4,20], and aromatic compounds such as benzene [21, 23], pyrrole and pyridine [23] and other N-containing heterocycles [24]. DFT underestimates the interaction energies between CO2 and electron donor systems [24]. However, it is possible that the use of functionals such as M06-2X [25] which accounts for some dispersion; or the addition of dispersion corrections as the ones proposed by Grimme et al. [26,27] may give results of enough quality to begin drawing correlations. The performance of M06-2X and B3LYP + D3 in various instances has been assessed showing good results [28]. We are trying to find not only a good candidate for further tests in their capacity of CO2 adsorption, but also find some of the physical origins of the terms that may contribute to the stabilization of the CO2 complexes.
Computational Methods
The density functional theory (DFT) approximation [29] as implemented in Gaussian 09 [30] was used for all the calculations. These were performed using the 6-311g + (d,p) basis set. Two functionals were employed: M06-2X [25] and B3LYP[31-33] including Grimme's D3 dispersion correction [26]. For comparison, additional calculations at the MP2/6-311g + (d,p) level were also carried out. Full geometry optimizations without symmetry constraints were carried out for all the stationary points. Harmonic frequency analysis allowed us to verify the optimized minima. The local minima were identified when the number of imaginary frequencies is equal to zero.
The interaction energies were determined as the difference between the energy of the CO2∙Heterocyle complex and the energies of the individual CO2 and nitrogen heterocycle [34,35]:

The zero-point energy (ZPE) of each species was considered to calculate the respective ΔZPE. Additionally, to account for basis-set superposition error (BSSE), use was made of the counterpoise correction (CPC) [36] as implemented in Gaussian. Fig. 1 contains the schematic representation of the nitrogen heterocycles considered in this study, which are imidazole (Im), 2methylimidazole (2-MeIm), benzimidazole (BzIm), and pyrazine (Pyz).

Two initial orientations were considered. Fig. 2 shows a schematic representation with imidazole as example. One of them featured an in-plane interaction between the CO2 and one nitrogen atom of the heterocyclic molecules (Fig. 2a). The other orientation contemplated a top-on interaction (Fig. 2b).

For all the complexes, a NBO [37] analysis was performed in order to calculate the resulting NBO charges of the CO2 and heterocyclic fragments to obtain a resulting charge transfer, Δq, from the heterocycle to CO2.
Results and Discussion
In order to keep a systematic presentation of the results, the in-plane complexes will be discussed first. The resulting optimized geometries, calculated with the M06-2X functional, of the in-plane complexes of CO2 with imidazole, 1; 2-methylimidazole, 2; benzimidazole, 3; and pyrazine, 4 are presented in Fig. 3.

The values of ΔZPE calculated for each of the in-plane complexes, the interaction energies, both the uncorrected, Eint, as well as those calculated with the counterpoise corrections with the addition of ΔZPE, Eint(CPC + ZPE), are presented in Table 1. The results for the MP2 calculations are presented in Table S1 of the Supplementary Material. Provided that in this particular study, the main interest falls on the determination of energetic trends, it can be seen that independently of the functional or level of theory used (DFT or MP2), the trend in the ordering for all the Eint and Eint(CPC + ZPE) values does not change. Thus, the relationship between the calculated values of -Eint(negative of Eint), for the in-plane complexes is
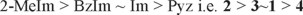
The M06-2X functional gave slightly more negative values (smaller than 2kJ/mol) compared to those obtained with B3LYP + D3. In addition, the interaction energies calculated at the MP2/6-311 + g(d,p) level are around 11 kJ/mol less negative than their DFT counterparts (Table S1, Supplementary Information) are. It can be seen that the uncorrected values for DFT and MP2 are very similar, but ΔZPE and BSSE are larger for MP2. The strongest and weakest interaction energies take place with the 2-methylimidazole (2) and pyrazine (4) complexes, respectively. The Eint values for the complexes with benzimidazole (3) and imidazole (1) are calculated to be very similar.
Table 2 contains some geometrical parameters of the complexes such as the O-C-O angle in CO2, the distance between one nitrogen atom in the heterocyle and the carbon atom of the CO2 molecule (N3-CCO2 distance), and the shortest distance between an H atom in the heterocycle and the oxygen of OCO2 (H-OCO2 distance). The results in Table 2 indicate that the O-C-O angle of the CO2 molecule for the in-plane complexes, experiences a change from 180° in the isolated molecule to an average of 176.1° ± 0.5°. As one way to assess the occurrence of Lewis acid-base interactions for the in-plane complexes, the respective calculated NBO charge transfers (Δq) from the heterocycle ring to CO2 are also presented in Table 2. Table S2 in the Supplementary Material contains the geometrical data for the MP2 calculations, it can be seen that the same trend as for the DFT calculations holds. Since the values of Eint for MP2 are less negative, consequently the distances are slightly longer.

In addition, to discuss the influence of the electrostatic interactions in the distortion of the C-O-C angle and on the values of Eint, Table 3 shows the electrostatic surface potential (ESP) charges calculated on the isolated heterocycles at the N3 site, and at the H atom that appears closer to CO2 in the complexes. The electrostatic potential maps for all the nitrogen heterocycles considered here are presented in Fig. 4. Following an order from the most negative (or positive, for H) to the least negative (or positive) the trend in the ESP charges is:



Table 2 indicates that the largest C-O-C distortion occurs with the 2-MeIm (2) complex (angle of 175.5°) which also corresponds to the largest value of charge transfer (Δq = -0.00981e) from the heterocycle to CO2. This complex also has the shortest N3-CCO2 distance among all the complexes although the ESP charge on N3 is the second most negative. However, the H charge is the largest positive (Table 3). Consequently, the H-OCO2 distance is the shortest. As shown in Table 1, the complex with 2-methylimidazole has also the strongest interactions, i.e. the largest Eint (in absolute value). In the case of the complexes with pyrazine (4) and imidazole (1), Table 1 shows that the weakest interactions occur with the pyrazine complex, followed by imidazole. In this case, while their calculated Δq values are both around -0.0091e, the C-O-C angle for the pyrazine complex is the least distorted (176.7°). Their N3-CCO2 distances are very similar (average of 2.71 ± 0.01 Å) although the charge on N3 in imidazole is slightly more negative, thus favoring a stronger electrostatic interaction for the Im complex compared to the pyrazine one. Moreover, the positive charge on H for imidazole is the larger of the two, influencing the H-OCO2 distance and the electrostatic interactions. Finally, the calculated Δq for the BzIm complex, 3, is slightly the smallest of all (-0.00876e), while its C-O-C angle (176.1°) is comparable to that in the complex 1 with imidazole. Comparing the results in Table 3, the charges on H are almost of comparable magnitude, but the smaller positive charge on BzIm also correlates with a larger H-OCO2 distance (Table 2). However, a larger negative charge on N3 for benzimidazole leads to a shorter N3-CCO2 distance, and consequently to a stronger interaction as seen in Table 1. The discussion above suggests a greater influence of the electrostatic interactions in the distortion of the C-OC angle for the in-plane complexes, as well in their interaction energies. Arnold et al. [38] established during their studies on the anions X-CO2 (X = I, Cl, Br) that distortion can come about mainly from electrostatic effects from the attractive X-/C and repulsive X-/O interactions but that charge transfer does occur as well and plays a role in the anion geometry. In the complexes under study, both interactions N3/C and H/O are attractive. It has also been stated that the interaction of CO2 with electron donors that causes the population of the LUMO will also cause a distortion of CO2 from linearity [39,40]. Additionally, Freund and Roberts argued [40], on the basis of the Walsh diagram of CO2, that the occupation of the 2πu-6a1 molecular orbital is important in determining the bond angle because this orbital is the one for which the bent molecule is strongly favored. The orbital 2πu corresponds to the LUMO of linear CO2 in the Walsh diagram. This diagram is included as Fig. S1 in the Supplementary Material. Thus, while the electrostatic interactions play an important role in the bending of CO2 for the in-plane complexes, it could also be expected that the charge transferred from the ring to CO2 would occupy the corresponding LUMO of CO2 and contributing to the electronic stabilization of the complex. Further, the charge transfer could be connected to Lewis acid-base interactions.
Moving on now to the discussion of the top-on complexes, some new materials with formula TL2[Ni(CN)4] (T = Ni2+, Co2+ or Mn2+ ; L = Im, BzIm, 2-MeIm, Pyz) have been recently synthesized and characterized [15-17], as mentioned in the Introduction. In those solids, the L ligands are located in between quasi-parallel T[Ni(CN)4] layers. Moreover, the coordination of the ligand to the T2 + metal is via the lone pair of the nitrogen atom of the heterocycles. Considering these new materials, where the adjacent layers would obstruct any CO2∙N in-plane interaction, it is worthwhile to describe the top-on complexes.
Fig. 5 shows the optimized geometries calculated with the M06-2X functional of the complexes formed between CO2 and imidazole, 1t; 2-methylimidazole, 2t; benzimidazole, 3t; and pyrazine, 4t.
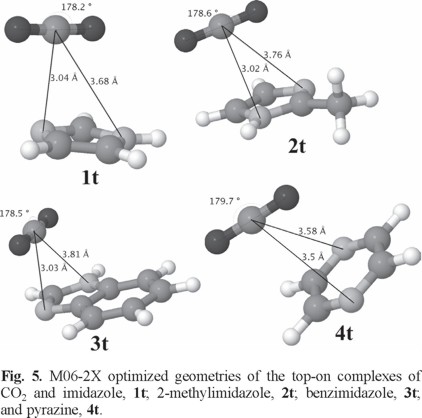
Table 4 shows the values of ΔZPE calculated for the top-on complexes, the interaction energies; both the uncorrected, Eint, as well as those calculated with the counterpoise corrections with the addition of ΔZPE, Eint (CPC + ZPE).
Here, the corrected values for the M06-2X functional gave more negative values of around only 1 kJ/mol with respect to the corresponding B3LYP + D3 results. Moreover, the interaction energies calculated at the MP2/6-311 + g(d,p) level are around 9 kJ/mol less negative than their DFT counterparts are. The results for the MP2 calculations are presented in Table S3 of the Supplementary Material. In line with the observations of Vogiatzis et al. [24], while the in-plane complexes are more stable, both arrangements are, in principle, favored (i.e. the Eint is negative in all DFT results). A point to note appears upon examination of the corrected results for the MP2 calculations, which indicate a very small positive value for the complexes with pyrazine and imidazole. Taking into account that this study is concerned in the drawing of energetic trends, more demanding and accurate calculations are set as an outlook. Nevertheless, once more the trend in the calculated values does not change, independently of the functional/level of theory used. The tendency of the calculated negative of Eint is in all cases

Comparison of these values with those in Table 1, it can be seen that the interactions between the heterocycles and CO2 are weaker than those taking place within the in-plane complexes.
Table 5 shows some geometrical parameters of the top-on complexes such as the O-C-O angle in CO2, the distance between CO2 and some selected atoms of the heterocyle, and the distance "d" defined as the distance between the plane of the heterocyclic ring and CCO2. The corresponding results for the MP2 calculations are presented in Table S4.

The O-C-O angle in all the top-on structures reveal a smaller deviation from linearity with respect to the in-plane species. In addition, the calculated Δq is around one third smaller with respect to the values for the in-plane complexes. The complexes with imidazole and its derivatives (1t, 2t, and 3t) have all almost the same O-C-O angle of around 178.55 ± 0.05°. For the pyrazine complex, 4t, the deviation from linearity is very small (angle of 179.7°).
For the top-on species, the orientation of CO2 rules out the type of Lewis acid-base/electrostatic interactions as described for the in-plane complexes. Proposing that the electrostatic interactions play a role for determining the distances between CO2 and the aromatic rings, it can be seen in Table 5 how in general the distances are larger, thus decreasing the contribution of electrostatic interactions. Further, the distances d for imidazole and its derivatives are almost identical since they range from 2.93 Å for benzimidazole to 2.96 Å for 2-methylimidazole (Table 5). However, their interactions energies are not very similar (Table 5). Only in the case of the pyrazine complex, the largest d (3.09 Å) corresponds to the least negative value of Eint. The DFT values of around -7 kJ/mol resembles that reported by Deshmukh et al. [41]. They calculated, at the cc-pVTZ/M06-2X level, a binding energy of -1.99kcal/mol for CO2 interacting with a pyrazine ring of one model with formula [Fe(CN4)2(Pyz)3]. Other weak interactions aside the electrostatic interactions associated with the interatomic distances should be taken into account. A point worth of notice is the fact that when the D3 correction was not used in combination with the B3LYP functional, the geometry optimization of the top-on configuration was not achieved in any way because in all cases the optimized geometries converged to an in-plane configuration. This points out towards the importance of the van der Waals interactions in the formation of the top-on complexes. Thus, the weaker van der Waals forces, along with the also weak dipole-quadrupole, and quadrupole-quadrupole interactions (discussed below) and the weaker electrostatic interactions could now be considered as the main contributing features in the formation of the top-on complexes. Accordingly, the top-on adducts are less stable than the in-plane species. For the case of pyrazine, any dipole (heterocycle)-quadrupole (CO2) interaction, is ruled out and only the quadrupole-quadrupole interactions can be considered. In fact, as it was mentioned before, the interaction energy for the pyrazine complex is the weakest amid all the top-on complexes. To assess the relationship between dipole moments and interaction energies, the dipole and quadrupole tensor moments of the isolated nitrogen heterocycles are listed in Table 6.
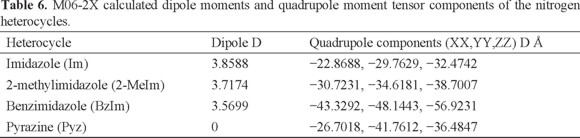
For the polar heterocycles, a direct correlation between the dipole moment of the heterocycle and Eint cannot be drawn because the largest absolute value of Eint is calculated for the complex with benzimidazole, which possesses the smallest dipole moment (Table 6). One additional interaction to be considered could be the weak charge donation from the aromatic ring to CO2, as it has been stated that the π-π interaction includes such type of charge transfer [23]. Nevertheless, by comparing the trends in the energies of interaction (Table 4) with the values of Δq (Table 5), it is clear once again that any direct correlation can not be established. The largest absolute value of Eint is calculated for the complex with benzimidazole, which possesses the smallest dipole moment and the smallest Δq. However, the distance d is the shortest (2.93 Å, Table 5). Additionally, the quadrupole moment tensor of benzimidazole is the largest among the aromatic rings. Thus, the interaction of the CO2 quadrupole with the heterocycle quadrupole is not negligible. Next, the second largest Eint corresponds to 1t, with a slightly larger d = 2.96 Å, but imidazole has the largest dipole moment, as well as the largest Δq, albeit the weakest quadrupole tensor components. Finally, the negative value of the calculated Eint for complex 2t with d = 2.95 Å is the smallest among all the imidazole-based complexes. In this case, 2 methylimidazole possesses a weaker dipole moment and a smaller Δq, although a slightly larger quadrupole tensor components, than imidazole. Therefore, it is clear that in the case of the top-on complexes, where the electrostatic interactions are weak, it is possible to outline some relations between the combination of electrostatic, dipole moment, quadrupole tensor components and Δq values.
Conclusion
According to the calculations presented here, for a given heterocycle under study, the in-plane orientation is more stable than the top-on one. In the first case, the contributions to the stabilization can be interpreted in terms of mainly electrostatic interactions plus Lewis acid-base interactions. The latter are related to the charge transfer Δq from the heterocycle ring to CO2. For the weaker top-on interactions, the use of a dispersion correction term, in this case Grimme's D3, added to the B3LYP functional proved essential to optimize the top-on complexes, highlighting the importance of the van der Waals interactions for the stabilization of those species. A high quadrupole tensor moment of the ring contributes significantly to the interaction energy when the complex is formed with an apolar species. In general, a combination of electrostatic, dipole-quadrupole, quadrupole-quadrupole and charge transfer interactions can be used to outline the trends in the interaction energies for the top-on complexes. In this study it was found that the in-plane and top-on complexes with pyrazine have the least favored energies of interaction compared with imidazole and its derivatives. With respect to a potential use as CO2 scrubbers, the materials containing imidazole and its derivatives are much better candidates for further experimental studies than those made with pyrazine.
Supplementary Material
Walsh diagram for CO2; Tables with the calculated Eint and selected geometrical parameters for the MP2 calculations for the in-plane and top-on species; and Cartesian coordinates of the in-plane and top-on geometries optimized with M06-2X.
Acknowledgments
Proyecto SEP-CONACYT CB 2011-01-166387, Proyecto ICYT-DF PICSO12-027. EHM acknowledges the Postdoctoral Scholarship from CONACYT.
References
1. http://www.epa.gov/climatechange/ccs/#important, accessed in June 2014.
2. House, K. Z.; Baclig, A. C.; Ranjan, M.; van Nierop, E. A.; Wilkox, J.; Herzog, H. J. Proc. Nat. Acad. Sci. U.S.A. 2011, 108, 20428-20433. [ Links ]
3. Rochelle, G. T. Science 2009, 325, 1652-1654. [ Links ]
4. Arstad, B.; Blom, R. Swang, O. J. Phys. Chem. A 2007, 111, 122-1228. [ Links ]
5. Pera-Titus, M. Chem. Rev. 2014, 114, 1413-1492. [ Links ]
6. Simmons, J. M.; Wu, H.; Zhou, W.; Yildirim, T. Energy Environ. Sci. 2011, 4, 2177-2185. [ Links ]
7. Sumida, K.; Rogow, D. L.; Mason, J. A.; McDonald, T. M.; Bloch, E. D.; Herm, Z. R.; Bae, T.-H.; Long, J. R. Chem. Rev. 2012, 112, 724-781. [ Links ]
8. Phan, A.; Doonan, C. J.; Uribe-Romo, F. J.; Knobler, C. B.; O'Keeffe, M.; Yaghi, O. M. Acc. Chem. Res. 2010, 43, 58-67. [ Links ]
9. Ruiz, E.; Alvarez, E.; Hoffmann, R.; Bemstein, J. J. Am. Chem. Soc. 1994, 116, 8207-8221. [ Links ]
10. Ruiz, E.; Novoa, J. J.; Alvarez, S. J. Phys. Chem. 1995, 99, 2296-2306. [ Links ]
11. Ruiz, E.; Alvarez, S. Inorg. Chem. 1995, 34, 3260-3269. [ Links ]
12. Culp, J. T.; Smith, M. R.; Bittner, E.; Bockrath, B. J. Am. Chem. Soc. 2008, 130, 12427-12434. [ Links ]
13. Culp, J. T.; Natesakhawat, S.; Smith, M. R.; Bittner, E.; Matranga, C.; Bockrath, B. J. Phys. Chem. C 2008, 112, 7079-7083. [ Links ]
14. Culp, J. T.; Madden, C.; Kauffman, K.; Shi, F.; Matranga, C. Inorg. Chem. 2013, 52, 4205-4216. [ Links ]
15. Gonzalez, M.; Lemus-Santana, A. A.; Rodriguez-Hernandez, J.; Knobel, M.; Reguera, E. J. Solid State Chem. 2013, 197, 317-322. [ Links ]
16. Gonzalez, M.; Lemus-Santana, A. A.; Rodriguez-Hernandez, J.; Aguirre-Velez, C. I.; Knobel, M.; Reguera, E. J. Solid State Chem. 2013, 204, 128-135. [ Links ]
17. Rodriguez-Hernandez, J.; Lemus-Santana, A. A.; Ortiz-López, J.; Jiménez-Sandoval, S.; Reguera, E. J. Solid State Chem. 2010, 183, 105-113. [ Links ]
18. Jorgensen, K. R.; Cundari, T. R.; Wilson, A. K. J. Phys. Chem. A 2012, 116, 10403-10411. [ Links ]
19. Hussain, M. A.; Soujanya, Y.; Sastry, G. N. Environ. Sci.Technol. 2011, 45, 8582-8588. [ Links ]
20. Cundari, T. R.; Wilson, A. K.; Drummond, M. L.; Gonzalez, H. E.; Jorgensen, K. R.; Payne, S.; Braunfeld, J.; De Jesus, M,; Johnson, V. M. J. Chem. Inf. Model. 2009, 49, 2111-2115. [ Links ]
21. Torrisi, A.; Mellot-Draznieks, C.; Bell, R. G. J. Chem. Phys. 2009, 130, 194703. [ Links ]
22. Torrisi, A.; Mellot-Draznieks, C.; Bell, R. G. J. Chem. Phys. 2010, 132, 044705. [ Links ]
23. Chen, L.; Cao, F.; Sun, H. Int. J. Quantum Chem .2013, 113, 2261-2266. [ Links ]
24. Vogiatzis, K. D.; Mavrandonakis, A.; Klopper, W.; Froudakis, G. E. ChemPhysChem 2009, 10, 374-383. [ Links ]
25. Zhao, Y.; Schultz, N. E.; Truhlar, D. G. J. Chem. Theory and Comput. 2006, 2, 364-82. [ Links ]
26. Grimme, S.; Antony, J.; Ehrlich, S.; Krieg, H. J. Chem. Phys. 2010, 132, 154104. [ Links ]
27. Grimme, S. WIREs. Comput. Mol. Sci. Chem. 2011, 1, 211-228. [ Links ]
28. (a) Valdes, H.; Pluhačkova, K.; Pitonák, M.; Řezáč, J.; Hobza, P. Phys. Chem. Chem. Phys. 2008, 10, 2747-2757. [ Links ] (b) Goerigk, L. Grimme, S. Phys. Chem. Chem. Phys., 2011, 13, 6670-6688. [ Links ] (c) Schneebeli, S. T.; Bochevaron, A. D.; Friesner, R. A. J. Chem. Theory Comput. 2011, 7, 658-668. [ Links ] (d) Zawada, A.; Kaczmarek-Kędziera, A.; Bartkowiak, W. J. Mol. Model. 2012, 18, 3073-3086. [ Links ] (e) Sedlack, R.; Janowski, T.; Pitoňák, M.; Řezáč, J.; Pulay, P.; Hobza, P. J. Chem. Theory Comput., 2013, 9, 3364-3374. [ Links ]
29. (a) Hohenberg, P.; Kohn, W. Phys. Rev. 1964, 136, B864. [ Links ] (b) W. Kohn, L. Sham. Phys. Rev. 1965, 140, A1133. [ Links ]
30. Gaussian 09, Revision D.01, Frisch, M. J.; Trucks, G. W.; Schlegel, H. B.; Scuseria, G. E.; Robb, M. A.; Cheeseman, J. R.; Scalmani, G.; Barone, V.; Mennucci, B.; Petersson, G. A. et al., Gaussian, Inc., Wallingford CT, 2009. [ Links ]
31. Becke, A. D. J. Chem. Phys.1993, 98, 5648-5652. [ Links ]
32. Lee, C.; Yang, W.; Parr, R. G. Phys. Rev. B. 1988, 37, 785-789. [ Links ]
33. Stephens, P. J.; Devlin, F. J.; Chabalowski, C. F.; Frisch, M. J. J. Phys. Chem. 1994, 98, 11623-11627. [ Links ]
34. Morokuma, K.; Kitaura, K. Energy Decomposition Analysis of Molecular Interactions. In Chemical Applications of Atomic and Molecular Electrostatic Potentials; Politzer, P., Truhlar, D. G., Eds.; Springer: New York, 1981; p. 216. [ Links ]
35. Jensen, F. Introduction to Computational Chemistry. 2nd ed.; John Wiley & Sons: England, 2007; 226-227. [ Links ]
36. Boys, S. F.; Barnardy, F. Mol. Phys. 1970, 19, 553-556. [ Links ]
37. Glendening, E.D.; Reed, A. E.; Carpenter, J. E.; Weinhold, F. NBO Version 3.1. [ Links ]
38. Arnold, D. W.; Bradforth, S. E.; Kim, E. H.; Neumark, D. M. J. Chem. Phys. 1995, 102, 3493-3509. [ Links ]
39. Aresta, M. Carbon Dioxide Reduction and Uses as a Chemical Feedstock, In Activation of Small Molecules; Tolman, W. B, Ed.; Wiley-VCH: Germany, 2006; p. 3. [ Links ]
40. Freund, H.-J.; Roberts, M. W. Surf. Sci. Rep. 1996, 25, 225-273. [ Links ]
41. Deshmukh, M. M.; Ohba, M.; Kitagawa, S.; Sakaki, S. J. Am. Chem. Soc. 2013, 135, 4840-4849. [ Links ]

