










Services on Demand
Journal
Article
 English (pdf)
English (pdf)
 Article in xml format
Article in xml format Article references
Article references
 Send this article by e-mail
Send this article by e-mailIndicators
-
 Cited by SciELO
Cited by SciELO -
 Access statistics
Access statistics
Related links
-
 Similars in
SciELO
Similars in
SciELO
Share

 Permalink
PermalinkJournal of the Mexican Chemical Society
Print version ISSN 1870-249X
J. Mex. Chem. Soc vol.58 n.3 Ciudad de México Jul./Sep. 2014
Article
Influence of EDTA on the Electrochemical Removal of Mercury (II) in Soil from San Joaquín, Querétaro, México
I. Robles,1 T. Serrano,1 J. J. Pérez,1 G. Hernández,2 S. Solís,2 R. García,3 T. Pi,4 and E. Bustos1,*
1 Centro de Investigación y Desarrollo Tecnológico en Electroquímica, S. C. Parque Tecnológico Querétaro, Sanfandila, Pedro Escobedo, 76703, Querétaro, México.Tel. +52 (442) 2-11-60-00. ebustos@cideteq.mx
2 Centro de Geociencias, Universidad Nacional Autónoma de México, Campus Juriquilla, Boulevard Juriquilla 3001, Querétaro 76230, México.
3 Centro de Ciencias de la Atmósfera, Universidad Nacional Autónoma de México, Ciudad Universitaria, 04510 México, D.F., México.
4 Instituto de Geología, Universidad Nacional Autónoma de México, Ciudad Universitaria, 04510 México, D.F., México.
Received January 21st, 2014
Accepted April 4th, 2014.
Abstract
The removal of mercury from soil and Ca-bentonite was performed using electrochemical treatment adding ethylendiaminetetra acetic acid (EDTA) as a complexing agent to improve the electrochemical removal of Hg (II) in soil from San Joaquín, Querétaro, México. During the electrokinetic treatment in the presence of 0.1 M EDTA, most of Hg (II) migrates toward the anode obtaining the highest removal efficiencies close to 70 % in bentonite after 9 h. Using 0.1M HCl only 65 % efficiency was attained after 13 h in the cathodic side. EDTA formed a negatively charged stable complex that migrates to the cathode by the application of the electrokinetic treatment across Hg - EDTA synthesized complex.
Keywords: Electrokinetic treatment, soil, bentonite, mercury, EDTA.
Resumen
La eliminación del mercurio en el suelo se llevó a cabo por tratamiento electrocinético usando EDTA como agente acomplejante para mejorar la eliminación electroquímica de Hg (II) en suelo de San Joaquín, Querétaro, México. Durante el tratamiento electrocinético en presencia de EDTA 0.1 M la mayor parte del Hg (II) migró hacia el ánodo con una eficiencia de 70 % después de 9 h en bentonita. Mientras que en el caso donde se utilizó HCl 0.1 M se obtiene un 65 % de remoción en 13 h del lado catódico. La presencia de EDTA forma un complejo estable que favorece la remoción de mercurio en el tratamiento electrocinético hacia el lado anódico a través del complejo sintetizado de Hg - EDTA.
Palabras clave: Tratamiento electroquímico, suelo, bentonita, mercurio, EDTA.
Introduction
Mercury is a transition metal of Group B, with the particularity that is the unique metal liquid at room temperature. The main source of Hg is cinnabar or mercury sulfide (HgS), which is a stable, insoluble compound, usually recovered as a sub-product during the processing of other minerals. One of the most important properties of Hg is its ability to dissolve many other metals forming amalgams such as silver,it was extensively used by dentists for many years and is currently discontinued due to their toxic effects [1 - 2]. Metallic mercury (Hg0) is primarily used for the production of chlorine gas and caustic soda and is part of some types of alkaline batteries, fluorescent lamps, electrical contacts, and instruments such as pressure gauges and thermometers [3]. Mercury in the environment can be present in elemental (Hg0), reduced (Hg2+ as mercuric, or Hg22+ as mercurous), or rented (methyl / ethyl mercury) chemical states. The mercuric and mecurous forms are more stable under oxidizing conditions [2]. In mildly reducing conditions, the organic or inorganic mercury can be reduced to its elemental form and be converted to alkylated by biotic or abiotic forms: these are the most toxic presentations of mercury as well as being soluble and volatile [2 - 3]. Hg (II) forms strong soluble complexes with a variety of organic and inorganic ligands in aqueous systems and soils. The Hg released into the air tends to settle and adhere to the organic matter present in soil [1 - 3].
The most commonly used techniques for the remediation of mercury contaminated soils have been classified as either excavation techniques or containment techniques, and are grouped as follows [4 - 7]: (a) ex situ treatments: physical separation, thermal treatments, hydrometallurgical treatments; (b) In situ recuperation: vapor extraction coupled with evaporation (soil), permeable reactive barriers; (c) In situ leaching and extraction: electrokinetic separation, interceptor systems, phytoremediation, passive remediation; (d) containment: pump and treat impermeable barriers, sealed surfaces and drainage, stabilization and solidification, sediment covering.
Electroremediation or electrokinetic treatment (EKT) has been successfully applied in a variety of soil restoration studies, this methodology has the advantage of exhibiting simultaneous chemical, hydraulic and electrical gradients. Indeed, for efficient mercury removal from a saturated soil using electroremediation treatments, the application of either an electric field or direct current through two electrodes (anode and cathode) is required. The electrodes are usually inserted in wells containing a supporting electrolyte made from inert salts, leading to improved electric field conductive properties [8].
Furthermore, since EKT is a physicochemical technique based on ion transport, it is an excellent tool for the removal of inorganic species, such as Hg+2 [9]. The main advantages of EKT, as compared with other soil treatment procedures, are [8]: (1) the electroosmotic flow is not dependent on either the pore or particle size, (2) the hydraulic gradient is enhanced by electromigration, (3) the treatment can be applied in situ, (4) it can be applied to low permeability soils, (5) there is minimal disruption of normal activities at the site, (6) the required investment is usually lower than that for other conventional treatments, and (7) it can be applied in conjunction with techniques such as pumping, vacuum extraction or bioremediation. The processes taking place during EKT can be classified into two main categories: (a) processes occurring as a consequence of the applied electric potential. These processes include electromigration, electroosmosis (mass transport), and electrophoresis (charged particle transport); (b) processes occurring in the absence of an electric potential. This includes concentration induced processes like diffusion, sorption, complexation, precipitation and acid - base reactions [7 - 10].
Besides promoting the removal of metals by electrochemical process, if a complexing agent is added, the latter is capable of forming a metal capture ligand. Metal elements tend to lose electrons during chemical reactions, which lead to metal ions, but these positively charged ions or cations do not exist in isolation but usually operate with negative ions, which are associated covalently to the metal center forming coordination compounds or complexes. The donor species (ligand) must have at least one pair of unshared electrons to form the bond [14 - 16]. Specifically, for mercury polluted soil electroremediation, the use of complexing agents like ethylendiaminetetra acetic acid (EDTA), KI, and NaCl under a constant potential gradient has been reported [11 - 13].
EDTA is a compound with four carboxyl groups and two amino groups, which can act as electron pair donors or Lewis bases. This compound has the capability of donate electrons and form coordinated covalent bonds with metal cations making one hexadentate ligand EDTA. However, in practice, usually EDTA is partially ionized, and therefore, less than six coordinated covalent bonds with metal cations are formed. The EDTA forms an octahedral complex with metal cations, mostly 2+, M2+, in aqueous solution [14 - 16]. Therefore, the objective of this work was to remove mercury from soil and clay by the electroremediation process facilitated by the use of EDTA as a complexing agent considering a real polluted soil from San Joaquin, Querétaro, México [15 - 16].
Methodology
Samples
Mercury - polluted soil from San Joaquín, Querétaro was used. The physicochemical characterization of this soil was previously performed. On the other hand, calcium bentonite was used as representative clay from soil [8 - 12, 17 - 19].
San Joaquin region lies in the south of the Sierra Gorda of Querétaro and is composed of approximately 172 square kilometers. It is located between 99°40' and 99°30' W and 21°10' and 20°50' N. Soil samples were taken from agricultural fields and mine tailings in the vicinity of two inactive mines. Site A was located at 436 661.00 m E and 2313568.00 m N, whereas site B was found at 440 384.60 m E and 2318085.71 m N. Human agricultural activities occur in proximity to both these sites. Five soil samples were taken and named as follows: (1) agricultural sample soil from site A, (2) former mine tailing sample that are now being used for agricultural activity from site A, (3) mine tailing sample from site A, (4) former mine tailing sample that are now being used for agricultural activity from site B, and (5) mine tailing sample from site B. For the first profile, sampling was performed at a depth of 10 cm, due to the high content of organic matter found in the area, which is from large plant development and high retention of nutrients available for crops [19 - 22].
Chemical analysis
In order to measure the metal concentrations in the soil samples, they were dried at room temperature for 3 days, and then sieved using a 2 mm mesh after weight amount of 0.5 g adding a mixture of HNO3 and HCl (1:3). MARS 5 analytical microwave oven to 110 psi for 30 min with a 14 - vessel capacity (HP-500), a pressure sensor (ESP-1500 Plus), and a temperature control system (EST-300 Plus) was used; after digestion, the solution was poured into a 25 mL volumetric flask and then deionized water was added to the mark and kept at 4 ºC until the analyses were carried out. The samples were analyzed by graphite furnace atomic absorption spectrometry (GFAAS) using a 932AA double beam GBC device, coupled with a 3000 graphite furnace accessory system, which consists on a GF3000 graphite power supply and a PAL3000 furnace auto sampler, both computer controlled. A deuterium lamp was used for background correction, while pyrolytically coated graphite tubes and boosted discharge hollow cathode lamps (Photron Super Lamp) were used for Cd, Fe and Pb analysis at 228.5, 248.3 and 217.0 nm respectively. Finally, a hollow cathode lamp (Photron) was used for Al, Cr, Mn, and V, at 309.3, 357.9, 279.5 and 318.5 nm respectively. Calibration standards were prepared with the same acid concentration as the samples. Cross check methods for standard additions were used. The detection limits, in µg⋅L−1 were 6.2, 0.07, 0.38, 2.04, 0.46, 0.78, 1.14 and 3.12 for Al, Cd, Cr, Fe, Mn, Ni, Pb and V, respectively. For the mercury analysis, we used a GBC model 932 atomic absorption spectrometer equipped with a GBC HG3000 continuous -flow automatic hydride generator and a GBC MC3000 Mercury Concentrator. Certified High-Purity Standards traceable to National Institute of Standards and Technology (NIST) were used for the determination of the metals.
The ions such as Na+, K+, Ca2+, and Mg2+ were analyzed using flame atomic absorption spectrometry with a GBC 932AA instrument at 589, 766.5, 422.7, and 282.5 nm, respectively. The Hollow cathode lamps (Photron) for these ions were used. Calibration standards were prepared from certified standards of each metal (high-purity standards traceable from NIST).
The ions of SO42−, NO3−, NH4+, and Cl− were analyzed using ion chromatography, following EPA method 300.1, using a Dionex ICS-2500 high-performance liquid chromatographer with a conductivity detector and a DionexIonPac AS14A column operating at a flow of 1 mL⋅min−1. Calibration standards were prepared from Dionex 7-Anion Standard, and data quality was evaluated using repeated analysis of certified standard IC-FAS-1A (Inorganic Ventures).
In addition to the elemental and ionic characterization, the pH, color, texture, organic matter content, cation exchange capacity, exchangeable ions, and soluble ions were also measured. Color was determined by comparing wet and dry samples with a Munsel color chart; the pH was determined by methods in water and potassium chloride. Textures were analyzed using the Bouyoucus method, which is based on separating particles, such as sand, silt, and clay, by their different densities. Organic matter content was determined by analyzing organic carbon content in soil samples using the Walkley and Black method. Cation exchange capacity was analyzed by saturating the soil with calcium ions and analyzing the capacity by titration, a method appropriate for neutral pH. Exchangeable and soluble ions were determined by analysis of a soil sample extract. These techniques were taken from NOM-021-SEMARNAT-2000 and ISRIC. All soil analyses were done in duplicate. Finally, the predominant crystallographic structures of the samples were examined using X-ray diffraction in a D8 Advance, Bruker AXS [21 - 22].
Quality control
Control samples on glass and plastic were prepared and analyzed. Control sample concentrations for trace metals were below equipment detection limits. Analysis precision and bias were determined with quality control check samples prepared in the laboratory. The limit of detection is defined as the concentration that produces a signal 3 times the standard deviation (SD) of the reagent blank readings. The uncertainty was calculated following the Eurachem / CITAC Guide (2000). The detection limits in µg⋅L−1 were Hg = 0.052, Al = 0.0261, Cr = 0.038, Fe = 0.953, Ni = 0.78, Cd = 0.007, Mn = 0.46, Pb = 1.14, and V = 0.263. The detection limits in mg L−1 were 0.22, 0.04, 0.31, 0.002, 0.006, 0.01, 0.002, and 0.05 for SO42−, Cl−, NO3−, Na+, K+, Ca2+, Mg2+, and NH4+, respectively.
Electrokinetic treatment
Calcium bentonite clay type (Merck brand) was previously polluted with 30 mg⋅Kg−1 mercuric chloride (HgCl2) of Merck 24 h before each experiment.
Electrokinetic treatment (EKT) was performed in the polluted soils and bentonite using 0.1 M HCl and 0.1 M EDTA for comparison. An acrylic rectangular cell (10 cm length × 2 cm width × 4 cm height) was used, as previously reported [14]. All EKT experiments were carried out using a PDC-GP 4303DU power supply. In the electrochemical cell, two clean and polish Ti plate electrodes were separated by 7 cm, which were the cathode and anode material. During the EKT experiments, the polluted soils and / or bentonite clay were placed between the two Ti plate electrodes in the electrochemical cell, then an electrical current was applied to facilitate the migration of Hg2+ ions toward the electrodes at different cell potentials: 0.4, 0.7, 1.0, 1.4, 1.7 and 2 V by 1 h each one. After selecting the best potential, different periods of time were evaluated: 1, 3, 7, 9, 11 and 13 h to verify the mercury removal across the time.
The content of mercury during EKT was evaluated using Anodic Stripping Voltammetry (ASV) with a pre-concentration potential of -0.6 V, a deposition time of 6 min, quiet time of 30 s, and scan rate of 20 mVs−1. An increment in current signal was observed with the increase of the mercury concentration. All experiments were carried out at room temperature (25 ± 1ºC). Calibration curves for mercuric quantification were done with a R2 of 0.999 [14].
Results
Metallic and ionic metal contents are shown in Figures 1 and 2, according to the NOM-147-SEMARNAT/SSA1-2004, the permitted level of mercury in agricultural or residential soils is 23 mgKg−1. As can be seen in Figure 1, all studied samples contained high mercury levels, above the regulatory limits. Also, all samples showed high levels of aluminum and iron, but only at levels that do not harm the environment, although high concentrations of aluminum can cause acidic soil [21].

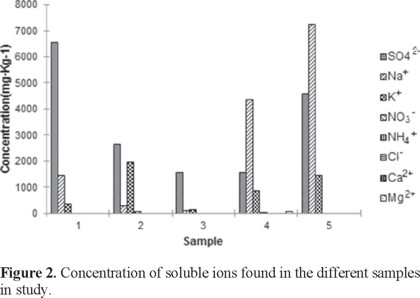
On the other hand, the results of analysis performed to determine the soluble ionic content (Figure 2) showed that the samples have high concentrations of sulfate, sodium, and potassium ions, depending on the source of the sample and the content of organic matter that increase the availability to cations, such as calcium, magnesium and ammonium, although found in small quantities, are of great importance because they compete with ionic mercury species (in soil, Hg2+) for exchange sites. Sulfate ions have a high affinity for oxidized (2+) metal ions, and the large number of mercury ions found in samples 1 and 5 can be associated to this ion (Figure 1).
Mercury concentration depended on the origin site of the sample. Samples from agricultural soil (samples 1 and 4, Table 1) had similar organic matter content and cation exchange capacities (CEC), whereas samples 2 and 3 had higher CEC. The mercury content found in samples 2 and 3 were similarly higher because the two characteristics (CEC and mercury) are related. Nevertheless, results from sample 5 did not show the same correlation but presented larger ionic concentrations of Ca2+, NH4+, and SO42− (Figure 2).
The high affinity between mercury and organic matter strongly influence the results of mercury speciation in soil environment. Humic substances make up to 50 % of the natural organic matter found in the soils and contained a high percentage of sulfur functional groups. The soluble fraction of these humic substances was composed of humic and fulvic acids, which are known to bind to Hg, forming compounds that dominate Hg speciation in soils [23 - 24].
Results from several field studies suggested that dissolved organic matter might reduce the bioavailability of mercury by complexation, thereby competing with the biota for the aqueous mercury. Humic substances have the potential ability to reduce Hg (II) to Hg (0) in soils as well as in aqueous systems. Mercury forms very strong complexes with humic substances and would thus be stabilized as Hg (II) in natural water. However, the formation of stable Hg-humic complexes also would lead to a subsequent reduction to Hg(0) by intramolecular electron transfer. The dissolved humic substances would be particularly important for the speciation and mobility of mercury in an aqueous environment, acting both as strong complexing agents and reducing agents, even when the biological transformation of Hg is slow [23 - 24].
The results of the granulometric determination (Figure 3) show the primary difference between samples, which is associated with its origin. Also, the number of available exchange sites depends on the texture of the soil because clay is known to have many of these exchange sites. Sand and silt are known to form aggregates where some metal compounds may remain adsorbed. The organic matter in the samples is located mainly in silts and clays because of its high surface area. Organic matter adds metals into its structure by carboxylic and −OH groups [23 - 25].
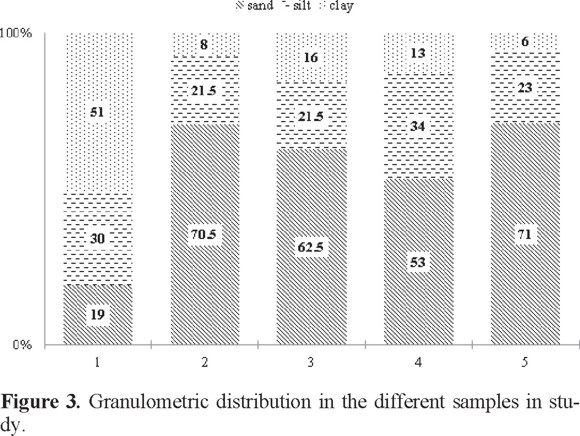
Adsorption of ionic species of mercury over mineral particles is attributed to the formation of strong bonds with the Oˉ functional groups that are found on the surface of minerals present in sand, clay, and silt. Complex forming reactions take place at the interface of the soil solution and the mineral surface, where surface charge and negative surface potential depend on the pH of the soil. The pH in the majority of natural soils was between 4 and 7, which is a consequence of the ionic content, although mercury species are more available in acid pH [23 - 25]. In this study, all of the samples had neutral pH, which means that the mercury found in the soil is chemically stable.
Cationic exchange capacity was determined by the amount of clay minerals and organic material present in the soil. The mercury adsorption mechanisms on clay lies in the cation exchange between soil particles because they contain exchangeable Ca2+, Mg2+, Na+, and K+. Mercury ions can displace monovalent cations on exchange sites [22 - 25].
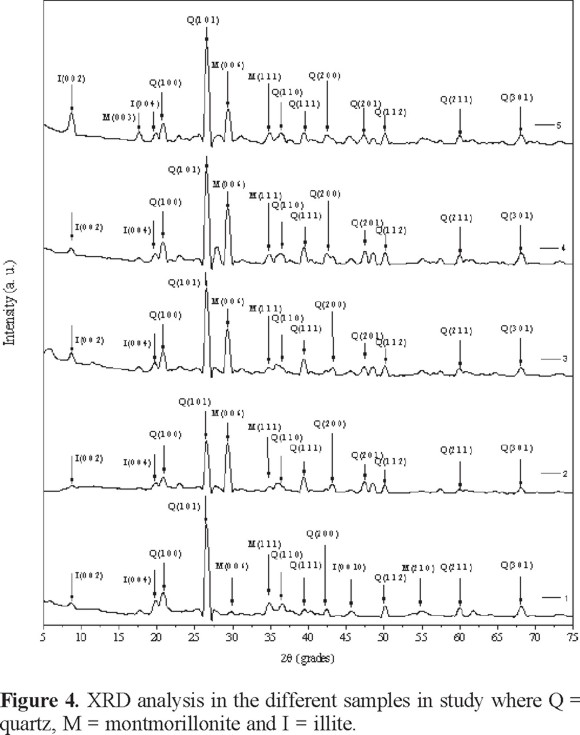
The mineralogical composition of the soil samples was obtained using X-ray diffraction (XRD) to qualitatively identify the crystalline phases present in the samples. XRD was also used to make a lattice parameter determination and a crystal structure analysis [23, 25].
Figure 4 shows the diffractogram for the soil samples, where the mineral found in greatest proportion was quartz, although the samples contained other structures, such as montmorillonite and illite, between 26.75° and 29.3° to 2ϑ. Quartz (SiO2; JCPS 046-1045 pattern) has a hexagonal lattice and a P3221 (154) symmetry group with a = 4.91344 and c = 5.40502, which make quartz a very strong structure and thus quite hard to cut. Montmorillonite (Na0.3(AlMg)2Si4O10OH26H2O; JCPS 012-0219 pattern) has a hexagonal lattice and a P101 symmetry group where a = 5.195 and c =17.93. Meanwhile, illite (KAl2(Si2Al)O10(OH)2; JCPS 43-0685 pattern) has a C2/c (15) symmetry group that has a base centered monoclinic lattice, where a = 9.017, b = 5.21, c = 20.43, a/b = 1.73071, and c/b = 3.92265. Montmorillonite and illite are not as hard as quartz, and these structures may experience ion exchange processes so that metals such as mercury can be added [24 - 25].
The physicochemical characterization helps to understand how mercury was deposited in the soil, in this way is important to know also how the chemical content affects the removal of mercury from the soil, and the physical analysis indicated how different particle size affect mercury caption. For this purpose, one soil sample (1) and Ca - bentonite was used to compare mercury removal efficiency using EDTA as a complexing agent to improve the treatment.
Initially, polarization curves were performed to find the conditions that facilitate the removal of mercury. During the experiment, the flow and pH was recorded every 15 min. The results indicated that the current increased proportionally to the applied potential for both polluted soil and clay (Figure 5). The soil pH near the anode and cathode varied between 3 and 4 in all the experiments.
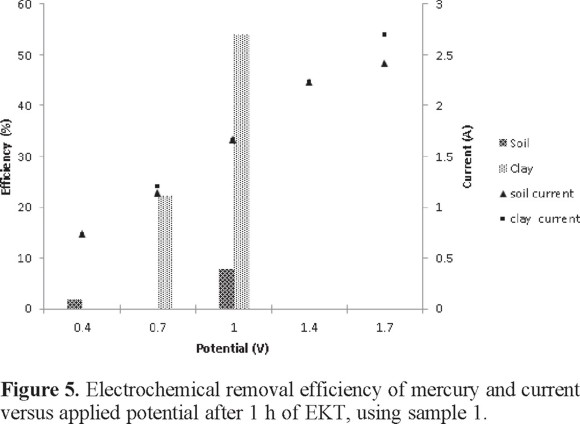
As seen in Figure 5, comparing the removal efficiency of mercury at different potential cell, the highest removal of mercury in soil and clay was obtained applying 1 V close to 60 % white a current close to 1.7 A in 0.1 M HCl. With this condition, the EKT of polluted soils and clay was conducted at different times of treatment: 1, 3, 7, 9, 11, and 13 h. During the experiments, the mercury removal, pH, and conductivity were measured, as shown in Tables 2 and 3 in the presence of 0.1 M HCl and 0.1 M EDTA, respectively.
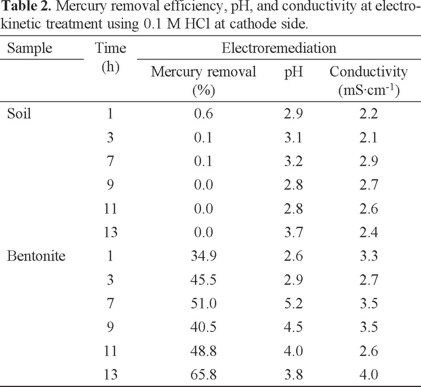
Comparing these results, the largest contribution was obtained for the bentonite sample, which showed the highest mercury removal close to 66 % (at the cathode) after 13 h of treatment, with pH close to 4 and 4 mScm−1 in the presence of 0.1 M HCl (Table 2), no significant efficiency was observed in the anode using this conditions, for this reason these results were omitted. A 70 % removal efficiency after 9 h of treatment was observed, with pH close to 4 and 11 mScm−1 in the presence of 0.1 M EDTA, in major proportion close to the anode than the cathode (Table 3). This result was important by the addition of the complexing compound, EDTA increased the mercury removal during EKT experiments than HCl by the presence of six donor atoms, which participated in the binding with the metal ion (Hg2+) and synthesized the complex Hg - EDTA in major proportion close to the anode due to the negative charge of EDTA (2-), which was directed by the electric field to the positive anode surrounding the mercury ion (Hg2+). This scenery is possible because the metal is located at the center of the complex, participating in coordination with both oxygen (carbonyl) and nitrogen (amino groups) of the EDTA framework. EDTA in the presence of potassium ions is the best extracting for removal of Hg+2 from soil via EKT as reported in the literature [14], decreasing the time of EKT from 13 h in presence of 0.1 M HCl to 9 h in presence of 0.1 M EDTA with similar efficiency removal close to 70 %.
Conclusions
Physicochemical properties of five contaminated samples from San Joaquín, Querétaro were determined. Mercury concentrations in the samples exceed the regulatory mercury concentration limit (23 mgKg−1) for agricultural and residential soils in México. In addition to high mercury concentration, these soils also have high concentrations of iron and aluminum, which affect the cation exchange capacity. High cation exchange capacity can also be attributed to the influence of sulfate, calcium, potassium, and sodium ions and to the content of organic matter.
Results of the physicochemical study showed a strong correlation between the number of available exchange sites and the amount of organic matter, as the functional groups in the organic matter can form stable bonds with metals. A higher percentage of clay also increased the number of exchange sites. Metallic ions with the higher affinity can displace ions such as calcium, magnesium, sodium, and potassium.
XRD of the main crystalline structures confirmed the presence of quartz and montmorillonite as the main crystallographic structures, both quartz and montmorillonite are capable of retaining large amounts of metallic ions. Quartz, in turn, can act as a surface where metal ions are adsorbed.
It is important to analyze soil properties, as soils with high toxic metal concentrations can adversely affect human health. Therefore, we propose that this place acts as a suitable site for treatment by the removal of mercury content, using a good alternative method, such as the application of electrokinetic treatment because of its previously mentioned properties.
After the EKT, the major mercury removal efficiency of 70.7 % was obtained in minor time (9 h) in presence of 0.1 M EDTA close to anode with pH 4.0 and 11.3 mS⋅cm−1, than in presence of 0.1 M HCl close to cathode after 13 h with pH 3.8 and 4 mS⋅ cm-1 (65.8 %). This result was evident by the synthesis of the complex Hg - EDTA and the removal of the pollutant across the pores between particles of soil from San Joaquín, Querétaro, México.
Acknowledgments
The authors would like to thank the Consejo Nacional de Ciencia y Tecnología de los Estados Unidos Mexicanos (CONACyT), the Academia Mexicana de Ciencias (AMC), the Fundación México-Estados Unidos para la Ciencia (FUMEC), the International Cooperation Program across bilateral cooperation Mexico - Hungary and Nanosciences and Nanotechnology Network with the project 194076 for the funding of this research. The authors thank to Prof. S. Rodil for English revision.
References
1. Yarto, M.; Gavilán, A.; Castro, J. La Contaminación por Mercurio en México Gaceta Ecológica, Secretaría de Medio Ambiente y Recursos Naturales. México, 2004, 72, 21-34. [ Links ]
2. Evaluación Mundial sobre el Mercurio, Programa de las naciones unidas para el medio ambiente, Ginebra Suiza, 2002. [ Links ]
3. Volke Sepulveda, T.; Velasco Trejo, J. A.; De la Rosa Pérez, D. A. Suelos Contaminados por Metales y Alternativas para su Remediación. Secretaría de Medio Ambiente y Recursos Naturales, Instituto Nacional de Ecología, México D. F. 2005. [ Links ]
4. Martínez, S. Data of Total Mercury Content in Soils and Plants in San Joaquín Querétaro, México, A Magyar Állami FöldtaniIntézet Évi Jelentése, 2010. [ Links ]
5. Stegmann, R.; Brunner, G.; Calmano, W.; Matz, G. Treatment of Contaminated Soil, Fundamentals Analysis Applications, Springer, Germany, 2001. [ Links ]
6. Reddy, K. R. Geotech. Geol. Eng. 2008, 15, 1. [ Links ]
7. García Rubio, A.; Rodríguez Maroto, J. M.; García Herruzo, F.; Vereda Alonso, C.; Gómez Lahoz, C. Limpieza de Suelos por Electrodescontaminación, Mejoras de la Técnica en el Movimiento de Contaminantes, Universidad de Málaga, 2000. [ Links ]
8. Huang, C. P., Cha, D., Chang, J.-H, Qiang, Z. Electrochemical Process for insitu Treatment of Contaminated Soils, Newark, Delaware, 2001. [ Links ]
9. Bustos, E. Remediación Electro-Cinética de Suelos Contaminados con Hidrocarburo. Chapter 5: Remediación de Suelos y Acuíferos Contaminados en México: Bases Teóricas y Experiencias Reales. Luis G. Torres Bustillos y Erick R. Bandala González. FUNDAP / CONCyTEQ / GTZ, México, 2013. [ Links ]
10. De la Rosa-Pérez, D. A.; Teutli-León, M. M. M.; Ramírez-Islas, M. E. Rev. Int. Contam. Ambient. 2007, 23, 129-138. [ Links ]
11. Reddy, K. R.; Camesselle, C. Electrochemical Remediation Technologies for Polluted Soils, Sediments and Grounwater, Willey, New Jersey, 2009. [ Links ]
12. Thöming, J.; Kliem, B. K.; Ottosen, L. M. Sci. Total Environ. 2000, 261, 137-147. [ Links ]
13. Reddy, K. Sep. Sci. Technol. 2005, 40, 1701-1720. [ Links ]
14. Robles, I.; García, M. G.; Solís, S.; Hernández, G.; Bandala, Y.; Juaristi, E.; Bustos, E. Inter. J. Electrochem. Sci. 2012, 7, 2276-2287. [ Links ]
15. Amrate, S.; Akretche, D. E. Chemosphere 2005, 60, 13761383. [ Links ]
16. Hajeb, P.; Jinap, S. J. Agric. Food Chem. 2012, 60, 6069-6076. [ Links ]
17. Porta, J. Edafología para la Agricultura y el Medio Ambiente, Ediciones Mundi-Empresa, México, 2003. [ Links ]
18. Martínez, S. Data of Total Mercury Content in Soils and Plants in San Joaquín Querétaro, México, A Magyar Állami FöldtaniIntézet Évi Jelentése, 2010. [ Links ]
19. Sposito, G. The Chemistry of Soils, Second Edition, Oxford University Press, 2008. [ Links ]
20. Tan, K. Environmental Soil Science, Marcel Dekker Inc., 2000. [ Links ]
21. Navarro, G. Química Agrícola, el Suelo y Los Elementos Químicos Esenciales para la Vida Vegetal, Mundi Prensa, 2003. [ Links ]
22. Pansu, M., Goutheyrow, J. Handbook of Soil Analysis: Mineralogical, Organic and Inorganic Methods, Springer, 2006. [ Links ]
23. Sjöblom, A.; Meili, M.; Sundbom, M. Sci. Total Environ. 2000, 261, 115-124. [ Links ]
24. Haitzer, M.; Aiken, G.; Ryan, J. Environ. Sci. Technol. 2003, 37, 2436-2441. [ Links ]
25. Introduction to Environmental Soil Physics http://www.sciencedirect.com/science/book/9780123486554 Daniel Hillel ISBN: 978-0-12-348655-4 accessed in January, 2014. [ Links ]

