










Services on Demand
Journal
Article
 English (pdf)
English (pdf)
 Article in xml format
Article in xml format Article references
Article references
 Send this article by e-mail
Send this article by e-mailIndicators
-
 Cited by SciELO
Cited by SciELO -
 Access statistics
Access statistics
Related links
-
 Similars in
SciELO
Similars in
SciELO
Share

 Permalink
PermalinkJournal of the Mexican Chemical Society
Print version ISSN 1870-249X
J. Mex. Chem. Soc vol.58 n.2 Ciudad de México Apr./Jun. 2014
Article
Synthesis, Chemical Structure Elucidation and Biological Studies on the Effect of Some Vital Metal Ions on Lisinopril
M. Zaky,1 Mohamed Y. El-Sayedl,1,2 Samy M. El-Megharbel,1,3 Sameh Abo Taleb,1 and Moamen S. Refat3,4
1 Department of Chemistry, Faculty of Science, Zagazig University, Egypt.
2 Faculty of Applied Medical Science, Al Jouf University-Al Qurayate.
3 Department of Chemistry, Faculty of Science, Taif University, 888 Taif, Kingdom Saudi Arabia.
4 Department of Chemistry, Faculty of Science, Port Said, Port Said University, Egypt. msrefat@yahoo.com
Received October 28th, 2013
Accepted February 12th, 2014
Abstract
Complexes of lisinopril as a pharmaceutical ligand with Ca(II), Mg(II), Zn(II), Fe(III) and VO(II), were synthesized and characterized by microanalysis, conductance, infrared and thermogravimetric (TGA/DTG and DTA) measurements. The lisinopril ligand acts as a monodentate feature via one carboxylate oxygen atom. Lisinopril and their complexes have been checked against some kinds of bacteria and fungi which gave a significant effect. The kinetic thermodynamic parameters such as: activation of E*, ΔH*, ΔS* and ΔG* were estimated using Coats and Redfern as well as Horowitz-Metzger equations.
Key words: Lisinopril complexes, Infrared spectra, electronic spectra, thermal analysis and antimicrobial activity.
Resumen
En este trabajo se sintetizaron los complejos de lisinopril como ligando farmacéutico con iones Ca(II), Mg(II), Zn(II), Fe(III) and VO(II). Los complejos obtenidos fueron caracterizados por microanálisis, mediante mediciones de conductividad, espectros de absorción IR y termogravimetría (TGA/DTG y DTA). El lisinopropil actúa como el ligando monodentado mediante el átomo de oxígeno del grupo carboxilo. La actividad biocida del lisinopril y sus complejos fue demostrada mediante ensayos con diferentes cepas de bacterias y micro hongos. Se estimaron parámetros cinéticos y termodinámicos tales como activación de E*, ΔH*, ΔS* y ΔG*, utilizando ecuaciones de Coats y Redfern y también de Horowitz-Metzger.
Palabras clave: Complejos de lisinopril, espectros de infrarrojo, espectros electrónicos, termoanálisis, actividad antimicrobiana.
Introduction
Metal ions are required for many critical functions in humans [1-5]. Scarcity of some metal ions can lead to disease. Well-known examples include pernicious anemia resulting from iron deficiency, growth retardation arising from insufficient dietary zinc, and heart disease in infants owing to copper deficiency [1]. Metals and metal complexes have played key role in the development of modern chemotherapy [6]. For example, anticancer platinum drugs appear in more chemotherapy regimens than any other class of anticancer agents and have contributed substantially to the success achieved in treating cancer over the past three decades. This is allowing the drug to be released in a controlled fashion or at specific location [7]. This approach may lead to the rescue of drugs that have failed because of poor pharmacology or high toxicity. For example, complexation of non-steroidal anti-inflammatory drugs to copper overcomes some of the gastric side effects of these drugs [8]. The release of cytotoxins such as nitrogen mustards from redox-active metals such as cobalt in the hypoxic regions of solid tumors has the potential to improve drug activity and reduce toxicity [9]. The metal based drugs are also being used for the treatment of a variety of ailments viz. diabetes, rheumatoid arthritis, inflammatory and cardiovascular diseases as well as diagnostic [10-12]. A number of drugs and potential pharmaceutical agents also contain metal-binding or metal-recognition sites, which were influence on their bioactivities [13-24]. Numerous examples of these metallodrugs and metallopharmaceuticals and their actions can be found in the literature, for instance: (a) several anti-inflammatory drugs, such as aspirin and its metabolite salicylglycine [13-16], suprofen [17], and paracetamol [18]; (b) the potent histamine-H2-receptor antagonist cimetidine [19] can form complexes with Cu2+ and Fe3+, and the histidine blocker antiulcer drug famotidine can also form stable complex with Cu2+ [20, 21]; (c) the anthelmintic and fungistatic agent thiabendazole, which is used for the treatment of several parasitic diseases, forms a Co2+ complex of 1:2 metal to drug ratio [22] (d) the Ru2+ complex of the anti-malaria agent chloroquine exhibits an activity two to five times higher than the parent drug against drug-resistant strains of Plasmodium faciparum [23]. However, it is known that some drugs act as potential ligands, a lot of studies are being carried out to ascertain how metal binding influences the activities of the drugs [24]. Metal-organic frameworks are not only stems from their tremendous potential applications in areas such as catalysis, molecular adsorption, magnetism, nonlinear optics, and molecular sensing, but also from their novel topologies and intriguing structural diversities [25-28]. On the other hand, many organic drugs, which possess modified pharmacological and toxicological properties administered in the form of metallic complexes [29], have the potential to act as ligands and the resulting metal-drug complexes are particularly important both in coordination chemistry and biochemistry [30-34] however, the study of metal-drug complexes is still in its early stages, thus representing a great challenge in current synthetic chemistry and coordination chemistry.
Lisinopril N2-[(1S)-1-carboxy-3-phenylpropyl]-L-lysyl-L-proline is a lysine analog of enalaprilat. It is a long-acting, nonsulfhydryl angiotensin-converting enzyme inhibitor (Formula 1), treatment of hypertension and congestive heart failure in daily dosages of 10-80 mg [35]. Pharmacological activity of lisinopril has been proved in previous experimental and clinical studies [36-37]. Historically, lisinopril was the third angiotensin-converting enzyme inhibitor (after captopril and enalapril) and was introduced into therapy in the early 1990. A number of properties distinguish it from other angiotensin-converting enzyme inhibitors: It is hydrophilic, has a long half-life and tissue penetration, and is not metabolized by the liver. Successful clinical trials in essential hypertension, renovascular hypertension and congestive heart failure have been conducted with lisinopril. Lisinopril is not significantly metabolized in humans; the absorbed drug is primarily excreted unchanged in urine. Peak serum concentrations of lisinopril are reached in about 6 h after administration. Mean maximum serum concentrations of lisinopril are about 80-140 ng/mL after a single oral dose of 20 mg [38]. Its indications, contraindications and side effects are as those for all angiotensin-converting enzyme inhibitors. Lisinopril is a subject of monograph in the United States Pharmacopoeia, which recommends an HPLC method for its analysis [39]. A variety of analytical techniques have been developed for the determination of lisinopril in pharmaceutical preparations, such as HPLC [40], GC [41], capillary electrophoresis [42-43], spectrophotometry [44-45], spectroflurimertric [40], micellar electrokinetic chromatography [46] and polarographic [45]. Techniques have been described for determination of lisinopril in biologic fluids including GC [47], HPLC [48], LC-MS [49], fluoroimmunoassay [50], radioimmuoassay [51], fluorozmatic assay [52] and ion selective electrodes [53].
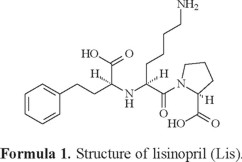
Lisinopril possesses are poor in electromagnetic absorbance due to week benzene chromophore; as a consequence, poor sensitivity can be achieved by UV spectrophotometric method. Therefore, most of these assays employ pre-column derivatization reactions to enhance the determination sensitivity. However, the preparation of derivatives is a time consuming procedure, and the derivatization frequently generates products that are unstable. Moreover, peak splitting owing to slow cis-trans isomerization may exist in the RP-HPLC [54]. GC and LC with mass spectrometric detection were widely applied to determination in biological fluids. In the literature survey, the synthesis and characterization of anti-hypertensive drugs complexes with some metal ions have been described [55-57]. The scope of this article is to discuss the coordination behavior of some of vital metal ions like Ca(II),Mg(II), Zn(II), Fe(III) and VO(II) with one of common blood pressure drug as lisinopril. The antibacterial and antifungal activities of lisinopril and its complexes were also evaluated.
Results and Discussion
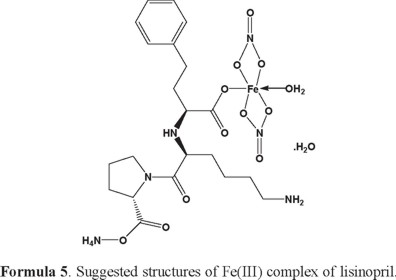
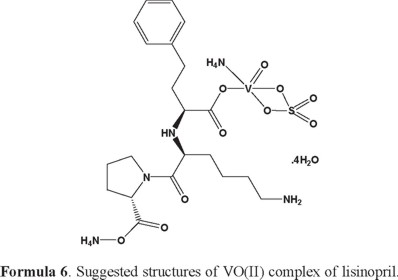
The reactions of lisinopril (Lis) with the metal ions Ca(II), Mg(II), Zn(II), Fe(III) and VO(II) gave a colored solid complexes in moderate to good yields (60-80%). The physical and analytical data, colors, percentage (carbon, hydrogen and nitrogen) and melting/decomposition temperatures of the compounds are presented in Table 1. The found and calculated percentages of elemental analysis CHN are in a well agreement with each other and prove the suggested molecular formula of the resulted lisinopril complexes. The complexes have high melting points (226-266) °C. The lisinopril ligand behaves as monodentate ligand and coordinates to the metal ions via one the oxygen atom of the carboxylate group. The isolated lisinopril complexes are 1:1 molar ratio of (M:Lis) where M=Ca(II), Mg(II), Zn(II), Fe(III) and VO(II).
Molar conductivities: The molar conductivity values for the Ca(II), Mg(II), Zn(II), Fe(III) and VO(II) complexes of lisinopril in DMSO solvent (1.00 × 10-3 M) were found to be in the range 5-27 Ω-1·cm2·mol-1 at 25 °C, suggesting them to be non-electrolytes [58] as shown in Table 1. Hence the molar conductance values indicate that no ions are present outside the coordination sphere so the Cl-, SO42- and NO3- ions may be exhibit inside the coordination sphere or absent. The obtained results were strongly matched with the qualitative analyses where Cl-, SO42- and NO3- ions are detected in case of Ca(II), Mg(II), Zn(II), Fe(III) and VO(II) complexes after degradation of these complexes by using nitric acid then precipitation of Cl- and SO42- by addition of AgNO3 and BaCl2 solutions, respectively to the solutions of the mentioned complexes were detected by using infrared spectral data. The low conductivity values [59] are in agreement with the low solubility of the above complexes in water, alcohol, chloroform, acetone and most of organic solvents. On other hand, they are soluble in DMSO, DMF and concentrated acids.
Electronic absorption spectra of lisinopril complexes: Electronic spectra of the lisinopril complexes were recorded in the 200-900 nm regions in DMSO. There are three detected absorption bands at (225 and 270) and 320 nm in the electronic spectrum of the free lisinopril ligand, these bands assigned to π-π* and n-π* transitions, respectively. These transitions occur in case of unsaturated hydrocarbons, which contain carbon atom attached with oxygen atoms as in carboxylic and ketone groups [60]. These bands of the free lisinopril ligand are bathochromically affected (red shifted) clearly in the electronic spectra of the Fe(III) and VO(II) complexes which show absorption bands at (259 and 325 shoulder) nm for Fe(III) complex and at (257 and 326 shoulder) nm for VO(II) complex which assigned to π-π* and n-π* transitions, respectively. This bathochromically change in the spectra of Fe(III) and VO(II) complexes indicated the association metal-to-ligand chelations. These results are clearly in accordance with the results of FT-IR and 1H-NMR spectra.
Infrared spectra of lisinopril complexes: The infrared spectral data of lisinopril and its complexes under investigation are recorded in Table 2. The spectra are similar but there are some differences which could give indication on the type of coordination. The infrared spectra of the free lisinopril show two broad bands observed at 3554 and 3396 cm-1, these bands are assigned due to the νas(OH) and νs(OH) stretching vibration of the -OH group. The infrared spectral bands of lisinopril complexes have a broad band's observed at 3400-3416 cm-1, these bands are assigned to the ν(OH) stretching vibration of the coordinated H2O molecules. The IR spectrum of lisinopril free ligand show strong absorption band at 1675 cm-1 due to ν(C=O) stretching vibration of the carboxylic group [61]. The Ca(II), Mg(II), Zn(II), Fe(III) and VO(II) complexes show no absorption band at 1675 cm-1 ν(C=O), that is indicative of the deprotonation of COOH group and involvement of the carboxyl group in the formation of M-O bonds [62]. The spectra of the complexes show two characteristic bands at (1404-1187), (1409-1187), (1400-1107), (1415-1155) and (1400-1105) cm-1 for Ca(II), Mg(II), Zn(II), Fe(III) and VO(II) complexes respectively, assigned as ν(COO-) asymmetric and symmetric stretching vibrations of the ligated carboxylate anion respectively. The criteria that can be used to distinguish between the three binding states of the carboxylate complexes have been studied previously by Deacon and Phillips [63]. These criteria are: (a) Δν > 200 cm-1 (where Δν =[ νas(COO-) - νs(COO-)]) this relation was found in case of monodentate carboxylate complexes, (b) bidentate or chelating carboxylate complexes exhibit Δν significantly smaller than ionic values (Δν < 100 cm-1), and finally, (c) bridging complexes show Δν comparable to ionic values (Δν~150 cm-1). Therefore, the difference value Δν is a useful characteristic for determining the coordination mode of the carboxylate group of the ligands. The observed Δν for Ca(II), Mg(II), Zn(II), Fe(III) and VO(II) complexes (Table 2) fall in the range 217-295 cm-1 indicating a monodentate coordination mode of the carboxylate group [64, 65]. The spectrum of lisinopril free ligand show absorption bands at 3100 and 3260 cm-1 due to ν(NH) stretching vibration of -NH and -NH2 groups respectively, these peaks are found in the spectra of the metal complexes with no significant change in their intensity and position. This observation suggests that both -NH and -NH2 groups haven't involved in the coordination. The ν(V=O) stretching vibration in the vanadyl complex is observed as expected band at 960 cm-1, which is a good agreement with those known for many vanadyl complexes [66]. The coordination of nitrato group for the Fe(III) ions were also supported by check the IR spectrum of the ferric(III) complex, where the nitrato complex displayed two starching vibration bands at 1350 cm-1 and 1100 cm-1 assigned to νas(NO2) and νs(NO2) respectively, the stretching motion of ν(N=O) is observed at 1415 cm-1 as a very strong band, while the two bending motion of the type Δ(NO2) are well resolved and observed at 790 and 697 cm-1 suggesting that bidentate nitrato group [67, 68]. Clearly the test against the presence of sulfate group in the VO(II) and Zn(II) complexes gave a positive result, this conclusion was supported by detected the two infrared frequencies bands at about 1100 and 600 cm-1 overlapping with angular deformation motions of the coordinated water molecules. New vibrating absorption bands were observed in the range of (550-615) cm-1 and (428-490) cm-1 are assigned to the stretching absorption bands of M-O and M-N bands, respectively [69]. According to the IR data, the lisinopril coordinated to the metal ions as monodentate ligand via the carboxylate oxygen atom [30].
1H-NMR of lisinopril and its complexes: The 1H-NMR data of free lisinopril and its Ca (II) complex, as an example are listed in Table (3). Upon comparison the free lisinopril ligand with its metal complex Ca(II), the significant change is the disappearance of the characteristic peak for hydrogen of -COOH at Δ = 11.0 ppm in Ca(II) complex indicates that the coordination of lisinopril ligand to Ca(II) through the deprotonated carboxylic group [70]. On other hand there is no significant change in the characteristic peaks of hydrogen for -NH, -NH2, -CH2 and -CH groups of aromatic ring in the free lisinopril ligand and its Ca(II) complex. This discussion is supported that these groups are not involved in the complexation [70]. The appearance of new peaks at 3.65 and 3.76 ppm in the spectra of Ca(II) complex which can assigned to the coordinated and uncoordinated of H2O molecules. In general, the structures of investigated complexes can be formulated as shown in Formulas (2-6) (3, 4, 5).
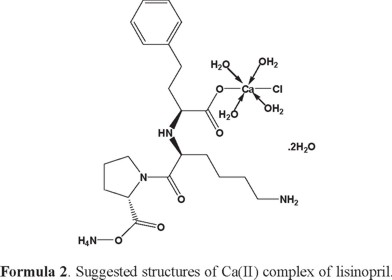
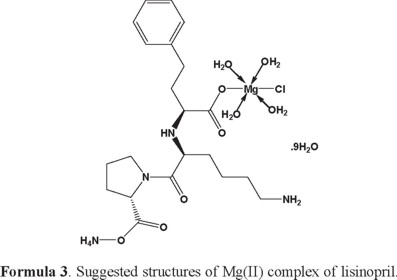
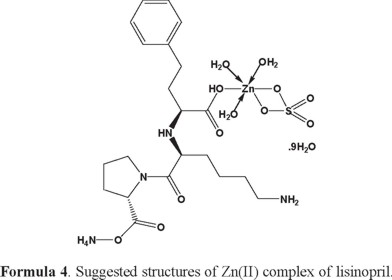
Thermal analysis: The obtained lisinopril complexes were studied by thermogravimetric (TG), differential thermogravimetric (DTG) and (DTA) analysis from ambient temperature to 800 °C under nitrogen atmosphere. The TG curves were redrawn as mg mass loss versus temperature and DTG curves were redrawn as rate of loss of mass versus temperature. The thermal decomposition results are summarized in Table 4.
[Ca(Lis)(Cl)(NH4)(H2O)4].2H2O complex: The thermal decomposition of Ca(II) complex of lisinopril with general formula [Ca(Lis)(Cl)(NH4)(H2O)4].2H2O is occurs at four steps. The first degradation step is in the range of 23-174 °C at DTGmax = 61 °C and its assigned to the loss of 4(H2O) molecules of hydration water with an observed weight loss 11.69% (calculated = 11.91%), the activation energy of this step is 25.3 kJmol-1. The second step is occurred within a temperature range 174-342 °C at DTGmax=296 °C due to the loss of 2(H2O) molecules of coordinated water + NH4Cl + C2H6 with a weight loss (Found = 20.57%, Calculated = 19.77%), the activation energy of this step is 75.9 kJmol-1. The third step is occurred within a temperature range 342-483 °C at DTGmax = 410 °C and DTA = 445 °C (exo) due to the loss C10H18N2O2 (organic moiety) with an observed weight loss 33.07% (calculated = 32.75%), the activation energy of this step is 120 kJmol-1. The fourth step is occurred within a temperature range 483-591 °C at DTGmax = 538 °C and DTA = 533 °C (exo) due to the loss of C3H5NO2 (organic moiety) with an observed weight loss 13.99% (Calculated = 14.39%), the activation energy of this step is 216 kJmol-1. The CaO + residual carbon atoms are remaining stable till 800 °C as final residual.
[Mg(Lis)(Cl)(NH4)(H2O)4].9H2O complex: The thermal decomposition of Mg(II) complex of lisinopril with general formula [Mg(Lis)(Cl)(NH4)(H2O)4].9H2O is occurs at four steps. The first degradation step is in the range of 54-196 °C at DTGmax = 92 °C and its assigned to the loss of 5.5(H2O) molecules of hydration water with an observed weight loss 14.13% (Calculated = 13.86%), the activation energy of this step is 37.1 kJmol-1. The second step is occurred within a temperature range 196-333 °C at DTGmax = 302 °C and DTA = 277 °C (exo) assigned to the loss of 7.5(H2O) molecules of hydration water with a weight loss (Found = 17.86%, Calculated = 18.89%), the activation energy of this step is 82.2 kJmol-1. The third step is occurred within a temperature range 333-460 °C at DTGmax = 399 °C and DTA = 332 °C (exo) assigned to the loss of NH4Cl + C7H13N (organic moiety) with an observed weight loss 23.39% (calculated = 23.02%), the activation energy of this step is 108 kJmol-1. The fourth step is occurred within a temperature range 460-601 °C at DTGmax = 510 °C and DTA = 513 °C (exo) due to the loss of C9H16N2O4 (organic moiety) with an observed weight loss 30.83% (calculated = 30.23%), the activation energy of this step is 188 kJmol-1. The MgO residual carbon atoms are remaining stable till 800 °C as final residual.
[Zn(Lis)(SO4)(NH4)(H2O)3].9H2O complex: The thermal decomposition of Zn(II) complex of lisinopril with general formula [Zn(Lis)(SO4)(NH4)(H2O)3].9H2O is occurs at five steps. The first degradation step is in the range of 30-127 °C at DTGmax = 81 °C and DTA = 82 °C (exo) and its assigned to the loss of 5(H2O) molecules of hydration water with an observed weight loss 11.09% (Calculated = 11.27%), the activation energy of this step is 55.3 kJmol-1. The second step is occurred within a temperature range 198-303 °C at DTGmax = 275 °C assigned to the loss of another 7(H2O) molecules of hydration water + C6H6 + NH3 gas with a weight loss (Found = 27.26%, Calculated = 27.68%), the activation energy of this step is 109 kJmol-1. The third step is occurred within a temperature range 303-443 °C at DTGmax=360 °C and DTA = 276 °C (exo) assigned to the loss of C5H12N (organic moiety) with observed weight loss 10.91% (calculated = 10.77%), the activation energy of this step is 77.5 kJmol-1. The fourth step is occurred within a temperature range 485-593 °C at DTGmax = 556 °C and DTA = 558 °C (exo) assigned to the loss of H2SO4 + C3H6N2O4 (organic moiety) with an observed weight loss 28.49% (calculated = 29.06%), the activation energy of this step is 296 kJmol-1. The fifth step is occured within temperature range 711-788 °C at DTGmax = 753 °C assigned to the loss of CH4 with an observed weight loss 2.28% (calculated = 2.00%). The ZnO + residual carbon atoms are remaining stable till 800 °C as final residual.
[Fe(Lis)(NO3)2(NH4)(H2O)].H2O complex: The thermal decomposition of Fe(III) complex of lisinopril with general formula [Fe(Lis)(NO3)2(NH4)(H2O)].H2O is occurs at three steps. The first degradation step is in the range of 43-138 °C at DTGmax = 73 °C and its assigned to the loss of 1.5(H2O) molecules of hydration water with an observed weight loss 3.86% (Calculated = 4.23%), the activation energy of this step is 49.9 kJ mol-1. The second step is occurred within a temperature range 138-311 °C at DTGmax = 250 °C and DTA = 278 °C (exo) assigned to the loss of 1/2(H2O) molecules of hydration water + NH4NO3 + C6H8NO3 (organic moiety) with observed weight loss 37.26% (calculated = 36.26%), the activation energy of this step is 69.1 kJ mol-1. The third step is occurred within a temperature range 311-462 °C at DTGmax = 407 °C and DTA = 416 °C (exo) assigned to the loss of C10H21N3O4 (organic moiety) with observed weight loss 38.59% (Calculated = 38.77%), the activation energy of this step is 111 kJ mol-1. The FeO residual carbon atoms are remaining stable till 800 °C as final residual.
[VO(Lis)(SO4) (NH4)2].4H2O complex: The thermal decomposition of VO(II) complex of lisinopril with general formula [VO(Lis)(SO4) (NH4)2].4H2O is occurs at four steps. The first degradation step is in the range of 49-122 °C at DTGmax = 76 °C and it is assigned to the loss of 2(H2O) molecules of hydration water with an observed weight loss 4.68% (calcd. = 5.34%), the activation energy of this step is 62.5 kJ mol-1. The second step is occurred within a temperature range 122-322 °C at DTGmax = 237 °C and DTA = 257 °C (exo) assigned to the loss of another 2(H2O) molecules of hydration + 2NH3 gas + H2SO4 + C3H4N (organic moiety) with observed weight loss 32.84% (Calculated = 32.94%), the activation energy of this step is 39.6 kJ mol-1. The third step is occurred within a temperature range 322-410 °C at DTGmax=362 °C and DTA = 357 °C (endo) assigned to the loss of C6H10O (organic moiety) with observed weight loss 14.88% (Calculated = 14.54%), the activation energy of this step is 130 kJ mol-1. The fourth step is occurred within a temperature range 410-528 °C at DTGmax = 476 °C and DTA = 478 °C (exo) assigned to the loss of C10H15N2O5 (organic moiety) with observed weight loss 36.47% (Calculated = 36.05%), the activation energy of this step is 219 kJ mol-1. The V + residual carbon atoms are remaining stable till 800 °C as final residual.
Kinetic studies: In the present investigation, the general thermal behaviors of the lisinopril complexes in terms of stability ranges, maximum temperature peaks and values of kinetic parameters, are shown in Table (5). The kinetic and thermodynamic parameters are evaluated using the Coats-Redfern and Horowitz-Metzger equations [71-73]. The entropy of activation, ΔS*, is calculated. The enthalpy activation, ΔH*, and Gibbs free energy, ΔG*, are calculated from ΔH* = E* - RT and ΔG* = ΔH* - TΔS*, respectively. The thermodynamic behavior of most lisinopril complexes with Ca(II), Mg(II), Zn(II), Fe(III) and VO(II) metal ions is non-spontaneous (more ordered) reactions (ΔS is negative value), endothermic reactions (ΔH > 0) and endergonic (ΔG > 0), during the reactions. The thermodynamic data obtained with the two methods are in harmony with each other. The correlation coefficients of the Arrhenius plots of the thermal decomposition steps were found to lie in the range 0.9637-0.9996, showing a good fit with linear function. The thermograms and the calculated thermal parameters for the complexes show that the stability of these complexes depends on the nature of the central metal ion. The thermal stability of the metal complexes was found to increase periodically with increase in atomic number of the metal and the larger value of charge/radius ratio [74].
Microbiological investigation of lisinopril complexes: Antibacterial and antifungal activities of lisinopril complexes were carried out against of bacteria as Escherichia coli (Gram -ve) and Staph albus (Gram +ve) as well as fungi as Aspergillus niger and Aspergillus flavus. The antimicrobial activities based on the size of inhibition zone around dishes were estimated. The free lisinopril has the lowest activity against four types of bacteria and fungi, while the Zn(II) complex was found to have the highest activity. The biological activities increase in the following order: Zn(II)/Lis > VO(II)/Lis > Fe(III)/Lis > Ca(II)/Lis > Mg(II)/Lis. The data are listed in Table 6.

Experimental part
Materials: All chemicals used in this investigation were of highest purity grade (Merck). Selected metal salts as CaCl2, MgCl2.6H2O, ZnSO4.H2O, Fe(NO3)3.9H2O and VOSO4.H2O were used. Lisinopril were received from Egyptian International Pharmaceutical Industrial Company.
Preparation of solid lisinopril complexes: [Ca(Lis)(Cl)(NH4)(H2O)4].2H2O (I), [Mg(Lis)(Cl)(NH4)(H2O)4].9H2O (II), [Zn(Lis)(SO4)(NH4)(H2O)3]. 9H2O (III), [Fe(Lis)(NO3)2(NH4)(H2O)].H2O (IV) and [VO(Lis)(SO4)(NH4)2].4H2O (V) were prepared, employing a 1:1 (metal:Lis) ratio. The complexes were prepared by mixing equal volumes (20 mL) of distilled water solution of CaCl2 (0.111 g, 1.0 mmol), MgCl2.6H2O (0.203g, 1.0 mmol), ZnSO4.H2O (0.180 g, 1.0 mmol), Fe(NO3)3.9H2O (0.404 g, 1.0 mmol) and VOSO4.H2O (0.163 g, 1.0 mmol) with a methanol solution of lisinopril (0.405g, 1.0 mmol). The mixtures are neutralized at pH (7.0-9.0) using 5% alcoholic ammonia solution, then the mixtures are heating at about ~60 °C for about one hour with stirring and left to evaporate slowly at room temperature overnight. The obtained precipitates were filtered off, washed several times by minimum amount of hot methanol and dried at 60 °C over anhydrous CaCl2. The melting points of the five lisinopril complexes are observed within the range of 226-266 °C. The Ca(II), Mg(II), Zn(II), Fe(II) and VO(II) complexes are soluble in dimethyl sulfoxide (DMSO) and N,N-dimethylformamide (DMF) with gently heating. The yield of lisinopril complexes is in the range of 60-75%.
Preparations of stock: Ten g of barium chloride dihydrate BaCl2.2H2O is weighted and dissolved in the least amount of distilled water. The volume was completed to 100 mL in a measuring flask to give 10% solution.
A weight of 0.1701 g of AgNO3 was dissolved in 100 mL distilled water in a dark measuring flask to obtain an approximate 0.01 M solution.
The stock solution of NH4OH was prepared by taking 15.15 mL of the concentrate NH3 (33% v/v) in 35 mL distilled water. The volume was then completed to 100 mL by methanol to give approximately (5% v/v) solution.
A weight of 0.2 g of the solid lisinopril complex was dissolved in the least amount of 2 mol nitric acid. The solution was evaporated near dryness. This process is repeated twice and the residue was dissolved in about 50 mL of hot distilled water to obtain a clear solution. The volume was completed to 100 mL in a measuring flask.
Apparatus and experimental conditions: Carbon, hydrogen and nitrogen content were determined using a Perkin-Elmer CHN Elemental Analyzer model 2400. The metal content was found gravimetrically by converting the compounds into their corresponding carbides or oxides. The Ca(II), Mg(II), Zn(II), Fe(III) and VO(II) contents were determined gravimetrically by the direct ignition of the complexes at 800 °C for 3 hours till constant weight. The residue was then weighted in the forms of metal oxides. Molar conductivities of freshly prepared 1.0 × 10-3 mol DMSO solutions of the complexes were measured using Jenway 4010 conductivity meter. IR spectra were recorded on Bruker FTIR Spectrophotometer (4000-400 cm-1) in KBr pellets. The electronic spectra were measured in the DMSO solvent with concentration (1.0 × 10-3 M) for the free ligands and their complexes using Jenway 6405 spectrophotometer with 1 cm quartz cell, in the range 200-900 nm. 1H-NMR spectra of the free ligands and their complexes were recorded on Varian Gemini 200 MHZ Spectrophotometer using DMSO-d6 as solvent and TMS as internal reference. Thermogravimetric analysis (TGA, DTG and DTA) was carried out in the temperature range from 25 to 800 °C in a steam of nitrogen atmosphere by using Shimadzu TGA-50 H thermal analyzer. The experimental conditions were: platinum crucible, nitrogen atmosphere with a 30 mL/min flow rate and a heating rate of 10 °C/min.
Most commonly used methods are the differential method of Freeman and Carroll [71] integral method of Coat and Redfern [72] and the approximation method of Horowitz and Metzger [73]. In the present investigation, the general thermal behaviors of the lisinopril complexes in terms of stability ranges, peak temperatures and values of kinetic parameters are discussed. The kinetic parameters have been evaluated using the Coats-Redfern equation:
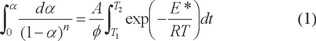
This equation on integration gives;
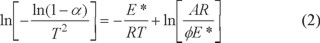
Where φ is the linear heating rate, R is the gas constant, T is the DTG temperature peak, α, is the fraction of the sample decomposed at time t, A is the pre-exponential factor. A plot of left-hand side against 1/T was drawn. E* is the energy of activation in J mol-1 and calculated from the slop and A in (s-1) from the intercept value. The entropy of activation ΔS* in (JK-1mol-1) was calculated by using the equation:
ΔS* = R ln(Ah/kB Ts) (3)
Where kB is the Boltzmann constant, h is the Plank's constant and Ts is the DTG peak temperature [75]. The Horowitz-Metzger equation is an illustrative of the approximation methods.
log [{1 - (1 - α)1-n}/(1 - n)] = E*θ/2.303RTs2 for n ≠ 1 (4)
When n = 1, the LHS of equation 4 would be log [-log (1 - α)]. For a first-order kinetic process the Horowitz-Metzger equation may be written in the form:
log [log(wα / wγ)] = E*θ/2.303RTs2 - log 2.303
Where θ = T - Ts, wγ = wα - w, wα = mass loss at the completion of the reaction. The plot of log[log(wα / wγ)] vs θ was drawn and found to be linear from the slope of which E* was calculated. The pre-exponential factor, A, was calculated from the equation:
E* /RTs2 = A/[ φ exp(-E*/RTs)]
The entropy of activation, ΔS*, was calculated from equation 3. The enthalpy activation, ΔH*, and Gibbs free energy, ΔG*, were calculated from; ΔH* = E* - RT and ΔG* = ΔH* - TΔS*, respectively.
Microbiological investigation: According to Gupta et al. [76], the hole well method was applied. The investigated isolates of bacteria and fungi were seeded in tubes with nutrient broth (NB) and Dox's broth (DB), respectively. The seeded (NB) for bacteria and (DB) for fungi (1 mL) were homogenized in the tubes with 9 mL of melted (45 °C) nutrient agar (NA) for bacteria and (DA) for fungi. The homogenous suspensions were poured into Petri dishes. The holes (diameter 0.5 cm) were done in the cool medium. A 100 μL of the investigated compounds were taken using a micropipette. After incubation for 24 h in an incubator at 37 °C and 28 °C for bacteria and fungi, respectively, the inhibition zone diameter were measured and expressed in cm. The antimicrobial activities of the investigated compounds were tested against of bacteria as Escherichia coli (Gram -ve) and Staph albus (Gram +ve) as well as fungi as Aspergillus flavus and Aspergillus niger. At the same time the pure solvent was tested with the antimicrobial investigations of the complexes. The concentration of each solution was 1.0 × 10-3 mol/L. Commercial DMSO was employed to dissolve the tested samples.
References
1. Sekhon, B.S. J. Pharm. Educ. Res. 2011, 2(1), 1-20. [ Links ]
2. Xiao, D.R.; Wang, E.B.; An, H.Y.; Su, Z.M.; Li, Y.G.; Gao, L.; Sun, C.Y.; Xu, L. Chem. Eur. J. 2005, 11, 6673-6686. [ Links ]
3. Drevensek, P.; Zupancic, T.; Pihlar, B.; Jerala, R.; Kolitsch, U.; Plaper, A.; Turel, I. J. Inorg. Biochem. 2005, 99, 432-442. [ Links ]
4. He, J.H.; Xiao, D.R.; Chen, H.Y.; Yan, S.W.; Sun, D.Z.; Wang, X.; Yang, J.; Yuan, R.; Wang, E.B. Inorg. Chim. Acta 2012, 385, 170-177. [ Links ]
5. Laxmi, K.; Sproules, S.; Pawar, O.; Markad, G.; Haram, S.; Puranik, V.; Salunke-Gawali, S. J. Mol. Struct. 2013, 1048, 223-229. [ Links ]
6. Gielen, M.; Tiekink, E.R.T., Eds., Metallotherapeutic Drugs and Metal-Based Diagnostic Agents, the Use of Metals in Medicine, Wiley, Chichester, 2005. [ Links ]
7. Bharti, S.K.; Singh, S.K. Pharmacia Lett. 2009, 1(2), 39-51. [ Links ]
8. Weder, J.E.; Dillon, C.T.; Hambley, T.W.; Kennedy, B.J.; Lay, P.A.; Biffin, J.R.; Regtop, H.L.; Daview, N.M. Coord. Chem. Rev. 2002, 232, 95-126. [ Links ]
9. Ware, D.C.; Brothers, P.J.; Clark, G.R. J. Chem. Soc. Dalton Trans. 2000, 925-932. [ Links ]
10. Nakai, M.; Sekiguchi, F.; Obata, M.; Ohtsuki, C.; Adachi, Y.; Sakurai, H.; Orvig, C.; Rehder, D.; Yano, S. J. Inorg. Biochem. 2005, 99, 1275-1282. [ Links ]
11. Chaviara, T.; Christidis, P.C.; Papageorgiou, A.; Chrysogelou, E.; Hadjipavlou-Litina, D.J.; Bolos, C.A. J. Inorg. Biochem. 2005, 99, 2102-2109. [ Links ]
12. Sadler, P.J.; Guo, Z. Pure Appl. Chem. 1998, 70, 863-871. [ Links ]
13. Baslas, R.K.; Zamani, R.; Nomani, A.A. Experientia 1979, 35, 455-456. [ Links ]
14. Gonzalez, B.E.; Daeid, N.N.; Nolan, K.B.; Farkas, E. Polyhedron 1994, 13, 1495-1499. [ Links ]
15. Nolan, K.B.; Soudi, AA. Inorg. Chim. Acta 1995, 230, 209-210. [ Links ]
16. Muller, J.G.; Burrows, C.J. Inorg. Chim. Acta, 1998, 275, 314-319. [ Links ]
17. Underhill, A.E.; Bougourd, S.A.; Flugge, M.L.; Gale, S.E.; Gomm, P.S. J. Inorg. Biochem. 1993, 52, 139-144. [ Links ]
18. Lawal, A.; Obaleye, J.A. Biokemistri 2007, 19(1), 9-15. [ Links ]
19. Kirkova, M.; Atanassova, M.; Russanov, E. Gen. Pharmacol. 1999, 33, 271-276. [ Links ]
20. Duda, A.M.; Kowalik-Jankowska, T.; Kozlowski, H.; Kupka, T.,J. Chem. Soc. Dalton Trans 1995, 2909-2913. [ Links ]
21. Kubiak, M.; Duda, A.M.; Ganadu, M.L.; Kozlowski, H. J. Chem. Soc. Dalton Trans 1996, 1905-1908. [ Links ]
22. Umadevi, B.; Muthiah, P.T.; Shui, X.; Eggleston, D.S. Inorg. Chim. Acta 1995, 234, 149-152. [ Links ]
23. Sanchez-del Grado, R.A.; Navarro, M.; Perez, H.; Urbina, J.A. J. Med. Chem. 1996, 39, 1095-1099. [ Links ]
24. Behrens, N.B.; Diaz, G.M.; Goodgame, D.M.L. Inorg. Chim. Acta 1986, 125, 21-264. [ Links ]
25. Xie, Y.-M.; Wu, J.-H. Inorg. Chim. Acta 2014, 412, 15-19 [ Links ]
26. Cao, X.; Yu, L.; Huang, R. J. Solid State Chem. 2014, 210(1), 74-78. [ Links ]
27. Sutrisno, A.; Huang, Y. Solid State Nuclear Magnetic Resonance 2013, 49-50, 1-11. [ Links ]
28. Zheng, X.-P.; Lu, Y.; Zhang, H.; Zhang, Z.-m.; Wang, E.-b. Inorg. Chem. Commun. 2013, 33, 29-32 [ Links ]
29. Lpez-Gresa, M.P.; Ortiz, R.; Perell, L.; Latorre, J.; Liu-Gonzalez, M.; Garcia- Granda, M.; Pe rez-Priede, M.; Cantn, E.; J. Inorg. Biochem. 2002, 92, 65. [ Links ]
30. Turel, I. Coord. Chem. Rev. 2002, 232, 27-47. [ Links ]
31. Xiao, D.R.; Wang, E.B.; An, H.Y.; Li, Y.G.; Xu, L. Cryst. Growth Des. 2007, 7, 506-512. [ Links ]
32. Xiao, D.R.; He, J.H.; Sun, D.Z.; Chen, H.Y.; Yan, S.W.; Wang, X.; Yang, J.; Yuan, R.; Wang, E.B. Eur. J. Inorg. Chem. 2012, 1783-1789. [ Links ]
33. Palumbo, M.; Gatto, B.; Zagotto, G.; Palu, G. Trends. Microbiol. 1993, 1, 232-235. [ Links ]
34. Sissi, C.; Andreolli, M.; Cecchetti, V.; Fravolini, A.; Gatto, B.; Palumbo, M. Bioorg. Med. Chem. 1998, 6, 1555-1561. [ Links ]
35. Lancaster, S.G.; Todd, P.A. Drugs 1988, 35, 646-669. [ Links ]
36. Rush, J.E.; Merrill, D.D. J. Cardiovasc. Pharm. 1987, 9, 99-107. [ Links ]
37. Gomez, H.J.; Smith, S.G.; Moncloa, F. Am. J. Med. 1988, 85, 35-37. [ Links ]
38. Beermann, B.; Till, A.E.; Gomez, H.J.; Hichens, M.; Bolognese, J.A.; Junggren, I.L. Biopharm. Drug Disposit. 1989, 10, 397-409. [ Links ]
39. The United States Pharmacopeia, 24th Revision, the National Formulary, 19th Revision, United States Pharmacopeial Convention, Inc., Rockville, MD, 2000. [ Links ]
40. El-Gindy, A.; Ashour, A.; Abdel-Fattah, L. J. Pharm. Biomed. Anal. 2001, 25, 913-922. [ Links ]
41. Qin, X.Z.; Nguyen, D.S.T.; Ip, D.P. J. Liq. Chromatogr. 1993, 16(17), 3713. [ Links ]
42. Kalal, H.S.; Rafiei, J.; Bani, F. Int. J. Environ. Res. 2010, 4, 289-296. [ Links ]
43. Du, J.; Ji, X.J.; Huang, H. Chin. J. Anal. Chem. 2009, 37, 681-684. [ Links ]
44. El-Yazbi, F.A.; Abdine, H.H.; Shaalan, R.A. J. Pharm. Biomed. Anal. 1999, 19(6), 819-827. [ Links ]
45. Razak, O.A.; Belal, S.F.; Bedair, M.M. J. Pharm. Biomed. Anal. 2003, 31, 701-711. [ Links ]
46. Bonazzi, D.; Gotti, R.; Andrisano, V. J. Pharm. Biomed. Anal. 1997, 16, 431-438. [ Links ]
47. Avadhamulu, A.B.; Pantulu, A.R.R. Indian Drugs 1993, 30, 646. [ Links ]
48. Qin, W.; Zhang, Z.; Tian, Y.; Xu, F. Biomed. Chromatogr. 2007, 21, 415-421. [ Links ]
49. Padua, A.A.F.; Barrientos-Astigarraga, R.E.; Rezende, V.M. J. Chromatogr. B 2004, 809, 211-216. [ Links ]
50. Yuan, A.S.; Gilbert, J.D. J. Pharm. Biomed. Anal. 1996, 14(7), 773-781. [ Links ]
51. Shepley, K.; Rocci, M.L.; Patrick, H. J. Pharm. Biomed. Anal. 1988, 6(3), 241-251. [ Links ]
52. Abdel-Fattah, L.; El-Kosasy, A.; Abdel-Aziz, L. J. Am. Sci., 2010, 6(10), 1115-1121. [ Links ]
53. Gustafsson, S.; Erriksson, B.M.; Nilsson, I. J. Chromatogr. 1990, 506, 75-83. [ Links ]
54. Barbato, F.; Morrica, P.; Quaglia, F. II Farmaco, 1994, 49, 457-460. [ Links ]
55. Hajnalka, J.; Pettinari, C.; Marchetti, F.; Kamu, E.; Nagy, L.; Troyanov, S.; Pellerito, L. J. Inorg. Biochem. 2003, 97, 370-376. [ Links ]
56. Anvarhusein, I.A.; Wazeer, M.I.M. Spectrochim. Acta Part A 2006, 65, 191-195. [ Links ]
57. Xiaoling, W.; Du, L.; Li, D.; Gong, Q.; Wang, L.; Lin, Y. Spectrochim. Acta Part A 2012, 94, 12-17. [ Links ]
58. Geary, W.J. Coord. Chem. Rev. 1971, 7, 81-122. [ Links ]
59. Kobelnik, M.; Cassimiro, D.L.; Santos, D.D.; Ribeiro, C.A.; Crespi, M.S. Chinese J. Chem. 2011, 29(11), 2271-2277. [ Links ]
60. Refat, M.S. Spectrochim. Acta Part A 2007, 68, 1393-1405. [ Links ]
61. Sadeek, S.A.; El-Didamony, A.M., El-Shwiniy, W.H.; Zordok, W.A. J. Arg. Chem. Soc. 2009, 97, 51-76. [ Links ]
62. Adam, A.A. J. Mat. Sci. Res. 2012, 1, 167-182. [ Links ]
63. Deacon, G.B.; Phillips, R.J. Coord. Chem. Rev. 1980, 33, 227-250. [ Links ]
64. Dendrinou-Samara, C.; Tsotsou, G.; Ekateriniadou, L.V.; Kortsaris, A.H.; Raptopoulou, C.P.; Terzis, A.; Kyriakidis, D.A.; Kessissoglou, D.P. J. Inorg. Biochem. 1998, 71, 171-179. [ Links ]
65. Nakamoto, K. Infrared and Ramman Spectra of Inorganic and Coordination Compounds, (4th ed.), New York: Wiley. pp. 230, 1986. [ Links ]
66. Bhattacharyya, S.; Mukhopadhyay, S.; Samanta, S.; Weakley, T.J.R.; Chaudhury, M. Inorg. Chem. 2002, 41, 2433-2440. [ Links ]
67. Nakamoto, K. Infrared Spectra of Inorganic and Coordination Compounds, Wiley Interscience, New York, 1978. [ Links ]
68. Ross, S.D. Inorganic Infrared and Raman Spectra, Mc Graw Hill, London, 1972. [ Links ]
69. Sharma, S.; Iqbal, S.A.; Bhattacharya, M. Orient. J. Chem. 2009, 25(4), 1101- 1104. [ Links ]
70. Refat, M.S. J. Mol. Struct. 2011, 985, 380-390. [ Links ]
71. Freeman, E.S.; Carroll, B. J. Phys. Chem. 1958, 62, 394-397. [ Links ]
72. Coats, A.W.; Redfern, J.P. Nature 1964, 201, 68-69. [ Links ]
73. Horowitz, H.W.; Metzger, G. Anal. Chem. 1963, 35, 1464. [ Links ]
74. Flynn, J.H.F.; Wall, L.A. J. Res. Natl. Bur. Stand. 1966, 70A, 487-523. [ Links ]
75. Wendlandt, W.W. Thermal Methods of Analysis, Wiley, New York, 1974. [ Links ]
76. Gupta, R.; Saxena, R.K.; Chatarvedi, P.; Virdi, J.S. J. Appl. Bacteriol. 1995, 78, 378. [ Links ]

