










Servicios Personalizados
Revista
Articulo
 Inglés (pdf)
Inglés (pdf)
 Artículo en XML
Artículo en XML Referencias del artículo
Referencias del artículo
 Enviar artículo por email
Enviar artículo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO -
 Accesos
Accesos
Links relacionados
-
 Similares en
SciELO
Similares en
SciELO
Compartir

 Permalink
PermalinkJournal of the Mexican Chemical Society
versión impresa ISSN 1870-249X
J. Mex. Chem. Soc vol.57 no.4 Ciudad de México oct./dic. 2013
Article
Submicrometric Fibrillar Structures of Codoped Polyaniline Obtained by Co-oxidation Using the NaClO/Ammonium Peroxydisulfate System: Synthesis and Characterization
Jorge Enrique Osorio-Fuente,1* Carlos Gómez-Yáñez,1 María de los Ángeles Hernández-Pérez,1 and Mónica de la Luz Corea-Téllez2
1 Departamento de Ingeniería en Metalurgia y Materiales, ESIQIE, Instituto Politécnico Nacional, UPALM, Av. Instituto Politécnico Nacional s/n, CP 07738, México, D.F. josorio@ipn.mx.
2 Departamento de Formación Básica, ESIQIE, Instituto Politécnico Nacional, UPALM, Av. Instituto Politécnico Nacional s/n, CP 07738, México D.F.
Received March 28, 2013.
Accepted June 3, 2013.
Abstract
A mixture of ammonium peroxydisulfate and sodium hypochlorite (NaClO) (co-oxidating system) were used to obtain polyaniline (PANi) doped with HCl and camphorsulfonic acid (CSA) (co-doping). The effect of HCl/CSA ratio added during polymerization on structure, morphology and electrical conductivity of the conducting polymer was investigated. When NaClO is used, the polymerization rate is substantially increased and the morphology changes from micrometric granular to nanometric fibrillar. CSA was used as complementary dopant but also to improve the solubility of PANi in common solvents. However, results suggest that quinone-like heterocycles containing carbonyl radicals as well as phenazine-type aromatic rings might be impeding an efficient doping in detriment of the conductivity.
Keywords: Polyaniline, Nanofibers, Codoping, Co-oxidant.
Resumen
El sistema de oxidación complementaria persulfato de amonio-hipoclorito de sodio (NaClO) se utilizó en la síntesis de polianilina (PANi) codopada con ácido clorhídrico y ácido canforsulfónico (ACS). Se investigó el efecto de la relación HCl/ACS presente durante la polimerización en la estructura, morfología y conductividad eléctrica del polímero conductor. Cuando se adiciona NaClO, la velocidad de polimerización aumenta notablemente y la morfología cambia de gránulos micrométricos a fibras en el rango de nanómetros. El ACS se empleó como dopante complementario y también para optimizar la solubilidad de la PANi en solventes comunes. No obstante, los resultados sugieren que los heterociclos quinonoides conteniendo radicales carbonilo así como anillos aromáticos tipo fenazina podrían obstaculizar un dopaje eficiente en detrimento de la conductividad.
Palabras clave: Polianilina, Nanofibras, Codopaje, Co-oxidante.
Introduction
Among semiconducting polymers, polyaniline (PANi) has attracted special interest due to its ease of synthesis, low cost, good optical and electrical properties added to its excellent environmental stability [1]. PANi was synthesized for the first time in 1862 [2] and it has been extensively studied since 1980 [3, 4]. This polymer has potential applications for electronic and optical devices such as LEDs, diodes, solar cells, sensors, rechargeable batteries and fuel cells [5-10]. Synthesis can be accomplished either by electrochemical or chemical oxidative techniques [11]. In the last case, polymerization is carried out in an aqueous solution of an oxidant and a strong acid that acts as dopant [12]. The final product of polymerization is the proton-ated emeraldine salt (ES) which is insoluble in common solvents as well as infusible, therefore, it is difficult to process by the techniques commonly applied to commercial polymers.
Several techniques [13] have been proposed in order to improve PANi solubility and hence, processability. One of these methods is the use of alkyl sulfonic acids, like dodecyl benzene sulfonic acid (DBSA) or camphorsulfonic acid (CSA) [14, 15]. The use of these acids has resulted in PANi with high conductivities, up to 102 S/cm. Also, the introduction of alkyl sulfonic acid groups in the main chain of the polymer enhances the solubility in common solvents such as benzene, chloroform, toluene or even in water [16]. However alkyl sulfonic acids are expensive reagents. Ruckenstein and Yin [17, 18] introduced codoping, i. e. simultaneous use of either DBSA or CSA along with a cheap acid like HCl showing that it is possible to obtain PANi with good conductivity and good solubility.
PANi conductivity can be improved even more by the manipulation of microstructural features. One-dimensional sub-micrometric PANi such as rods, wires, tubes, belts and fibers present better conductivities [19] when compared to particles showing irregular shapes which are commonly seen in PANi [20]. Moreover, fibrillar microstructures have potential applications as conducting polymeric molecular wires [21], superhy-drophilic and superhydrophobic devices [22], chemical sensors [23], actuators [24] and biosensors [25]. It has been observed that nanofibers are produced during the first stage of polymerization, even during the conventional synthesis [26]. However, when polymerization goes on, initial fibers evolve to form granules with an irregular morphology. The abundance of nanofi-bers is then, determined by polymerization kinetics [27]. Based on this idea, Li and Li [28] obtained PANi nanofibers, using sodium chlorite (NaClO2) instead ammonium peroxydisulfate ((NH4)2S2O8) (APS) as initiator in order to speed up the process suppressing the granule formation stage. Polymerization takes less of an hour, achieving conductivities up to 2.76 S/cm. In comparison, 5 hours were necessary to achieve the same value of conductivity by using APS as oxidant. Employing a mixture of sodium hypochlorite (NaClO) and APS, Rahy et al. [29] synthesized PANi nanofibers of few micrometers in length, and 40 nm or less in diameter, showing high conductivity (24.4 S/cm) and a narrow molecular weight distribution. This method has the advantage that it is possible to use commercial bleach instead of NaClO reagent grade to obtain PANi nanofibers.
The intention of this work was to produce PANi combining the advantages provided by CSA and HCl as dopants, and the mixtures of oxidants ((NH4)2S2O8 and NaClO)). The combination of dopants could control the morphology of PANi, besides the conductivity, whereas the polymerization rate could be controlled with the mixture of oxidants.
Results and Discussion
UV-VIS spectra of the polyaniline samples are shown in Fig. 1. In all cases, three peaks were observed; the first in the range of 350-360 nm, the second in 470-480 nm, and the third one in 845-850 nm for codoped PANi samples (Fig. 1a and Fig. 1b). For codoped - co-oxidized PANi samples the peaks are in 320-335 nm, 460-470 nm, and 840-850 nm (Fig. 1c and Fig. 1d). The region from 350 to 360 nm has been assigned to π - π* transition in the benzenoid structures [31] and the peak in the medium region has been assigned to polaron band transition of the PANi main chain [32]. These two peaks are overlapped in all cases, forming a shoulder, which is characteristic of PANi with HCl+CSA codoping [18]. The peak at the highest wavelength value has been attributed to the π to the localized polaron band transition of doped PANi [33]. As it is observed in Figure 1 this peak is wide and suggests that the sites, along the main chain where dopant ions can be allocated, are not saturated, that means there is a partial doping situation [34]. Highly doped PANi would show a very well defined and intense peak around 850 nm along with a free-carrier tail extending into the near-infrared region [34].
To determine the band gap, the following expression was used

Where ΔE is band gap energy, h is Planck's constant, c is velocity of light and λ is the wavelength corresponding to the π - π* transition [35]. The computed band gap values are listed in Table 1.
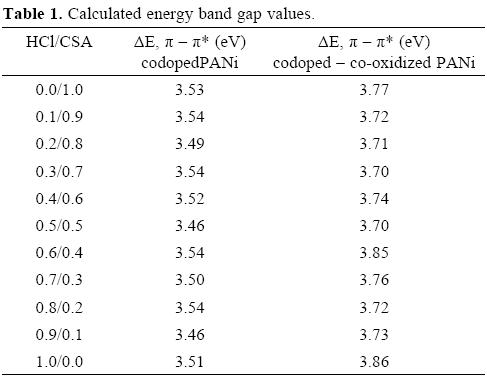
For PANi doped with alkyl sulfonic acids values of band gap are in the range of 3.88 - 4.51 eV [36] so the values shown in Table 1 are in good agreement with those previously reported. There is not a simple relationship between the Eg values and the HCl/CSA ratios in either the codoped or codoped-co-oxidized groups. However, when NaClO is used a clear increment in the Eg is observed in Table 1 which indicates that the effective conjugation π - π* decreases [36]. This increment has been related to a stretching in the atomic bonds along the PANi molecule [37]. NaClO is not only increasing the polymerization rate, it is promoting, possibly, the tail-to-tail coupling between the aniline radical cations during polymerization [38]. This kind of polymerization produces an increase in the torsion angle between the adjacent fenile rings [36, 37] by stretching the atomic bonds due to reaction through ortho position [38, 39] leading to further oxidation with eventual production of phenazine-type moieties along the backbone of the conducting polymer [40].
In sake of clarity Figure 2 shows some ATR-FTIR spectra of the as-synthesized PANi samples which were selected to show the main features. The wavenumbers of the observed peaks are listed in detail in Tables 2 and 3 and they also show the molecular structures that have been associated to the different peaks.
As observed in Table 2, the peak at 1740 cm-1 is not present for the sample with dopant ratio of 0.7/0.3. One possible explanation assumes that the carbonyl species causing the strong infrared absorption at 1740 cm-1 are those present in oligomers containing quinone-like heterocycles produced during polymerization [51].
On one side, when [CSA] < 0.3, the PANi molecule is doped with HCl and acquires a coiled conformation. CSA molecule cannot be attached to the former PANi molecule because closely conformation and bulky size of CSA molecule. The reactive site is not sufficiently exposed, slowing polymerization rate [18]. CSA is discarded during filtering process and due to the slow polymerization quinone-like oligomers are produced.
On the other side, when [CSA] > 0.3, there is enough CSA to dope the polymer main chain stretching, in this way, the molecule and making the CSA-doped macromolecules less coiled. However, these bulky CSA functional groups attached in the main chain impose a steric hindrance to the system also producing low weight chains by slowing polymerization rate [18] which favors, again, the quinone-like oligomer formation.
At [CSA] = 0.3 the molecules will be less coiled because CSA moieties, but enough reactive because they are more exposed and contain HCl-doped units which react more easily [17, 18]. Thus, polymerization rate is fast enough to produce long chains, preventing the production of oligomers containing quinone-like units. So the peak at 1740 cm-1 does not appear.
The peak at 1370 cm-1 has been assigned to the C=N stretching vibration mode in phenazine-type ring [41] and this peak is not present in the 0.7/0.3-0.4/0.6 codoped PANi samples (see Table 2). This absence suggests that in the former HCl/CSA ratios there is not phenazine-type aromatic ring production during polymerization. When values of wavelength are compared between Table 2 and Table 3, it is evident a slight red-shift of the peaks of one group with respect to the other. This red shift is related with transformation of quinone diimine structure to semiquinone radical cations [42].
Selected X-ray diffraction patterns are shown in Figure 3 and Figure 4. The patterns in Figure 3 present the peak at around 2θ = 25° which has been widely reported and it is assigned to the periodicity due to the stacking of polymer chains in the perpendicular direction of the chain axis [43]. According to Klug and Alexander [44] the interchain distance, R, is given by R = 5λ/8senθ, where λ is the wavelength of the X-ray radiation and 8 is the Bragg angle. In this case where 2θ = 25°, the interchain distance is in the range 4.33-4.43 Å, which is in agreement with values previously reported [36,45].
Two more peaks can been observed; one at around 15.4° and other at around 21.4°. These peaks along with that in 25° match with the pattern of an orthorhombic structure [46] reflecting in the planes (001), (010) and (100). Some codoped-cooxidized PANi diffraction plots (Fig. 4) show three additional peaks: at around 32, at 45.5 and 56.5 which coincide with those peaks observed by Rahy et al. [29]. When these additional reflections appear in the difractogram, the intensity of the peak at 25° diminishes and other two peaks are observed at around 22.5 and 28°. Former is related with loss of cristal-linity, involving predominance of insulating amorphous phase over crystalline conducting phase [47] and suggests that these polymers are constituted by low molecular weight, short chain oligoanilines [48].
SEM micrographs of codoped and codoped-cooxidized PANi are shown in Figure 5. The Fig. 5a shows a micrometric flake-like morphology, while in Figure 5b can be seen a morphology essentially formed by small aggregates. The aggregate size is ~0.35 μm. When coinitiators along codopants are used, morphology acquires a fiber-like structure, as can be seen on Figures 5c and 5d. From these pictures, one can find that the average diameter of the fibers in the two HCl/CSA ratios is ~80-100 nm for 0.4/0.6 ratio and ~60-90 nm for 0.2/0.8 ratio. In both cases, the average length of the fibrillar structures is found to be between 300 nm and 500 nm. Thus, higher CSA content promotes thinner fibers and a more uniform morphology. This can be attributed to the secondary growth of the initially formed nanofibers [47]. Average diameters of the fibers are wider than those reported previously in literature [28, 49, 50].
Fig. 6 presents curves of conductivity as a function of HCl/ CSA ratio for codoped PANi and codoped-cooxidized PANi pellets. Experimental error was determined to be of around ±0.022 S/cm (see experimental section), this value suggests that the trend in the conductivity measurements shown in Figure 6 is repeatable.
It was expected that some molecular and microstructural characteristics of PANi samples should influence strongly the conductivity. Codoped-cooxidized samples have a wider band gap, higher oxidation value and lower cristallinity as compared to the codoped samples which suggests that codoped-cooxi-dized samples should exhibit lower conductivity. However, these last samples also present fibrillar structure that should have some improvement on the conductivity with respect to the codoped samples.
These two contrary trends presented in codoped-cooxidized samples could explain why conductivity in both groups of samples is quite similar as observed in Figure 6. Still, conductivity values are rather lower than expected according to some reports [28, 29, 50]. It seems that additions of CSA or the use of NaClO are in detriment of conductivity because the sample oxidized only with APS and doped with HCl presented, by far, the highest conductivity. These results suggest that the factor controlling conductivity is the effective doping. That is, secondary reactions are, somehow, impeding the incorporation of dopant into the main chain. The production of different oligomers during polymerization of PANi has been observed [51, 52]. Among these oligomers, species containing carbonyl radicals, such as quinone-like heterocycles, have been reported to prevent acid doping [53].
Conclusions
(1) Polyaniline codoped with CSA and HCl, showing a nano fiber-like structure could be synthesized by the in situ chemical polymerization of aniline hydrochloride, using ammonium peroxydisulfate and sodium hydrochloride as coinitiators. (2) The morphology of codoped PANi changes from micrometric granular to nanometric fibrillar when sodium hypochlorite is used. (3) The use of sodium hypochlorite resulted in PANi samples with wider band gap as compared to the samples oxidized only with APS. (4) The results suggest that dopants are not incorporated into the main chain in efficient way in detrimental of conductivity due probably, to the presence of oligomers containing quinone-like heterocycles and phenazine-type aromatic rings.
Experimental
Reagents
Aniline hydrochloride (Fluka) was used as received. Ammonium peroxydisulfate (Sigma-Aldrich), sodium hypochlorite 15% (Sigma-Aldrich), hydrochloric acid (Fermont) and camphor-sulfonic acid (Fluka) were analytical grade and used without further purification. Distilled water was used in all experiments.
Synthesis
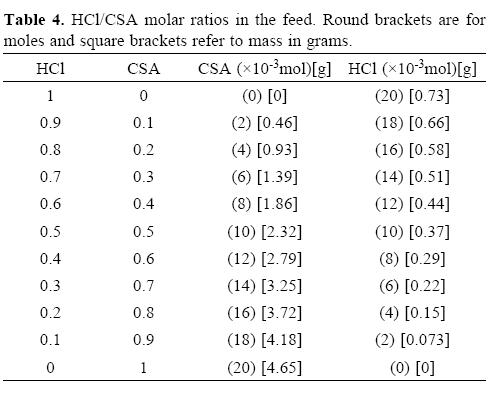
Polyaniline was synthesized according to the procedure described by Stejskal and Gilbert [30]: aniline hydrochloride (2.59 g, 20 mmol) was dissolved in 100 mL distilled water by magnetic stirring in a 250 mL beaker. Hydrochloric acid and camphorsulfonic acid in selected HCl/CSA ratios (in such a way that moles (CSA+HCl) / moles aniline hydrochloride = 1) were added, while stirring, to previous solution [18] (see Table 4). A solution of ammonium peroxydisulfate (APS) (5.71 g, 25 mmol) in 100 mL distilled water was added slowly, without stirring, to the former aniline mixture, followed by quick addition of 25 mL of an aqueous sodium hypochlorite (5 wt%) solution [29], being APS to NaClO molar ratio equivalent to 1.18:1. It was considered that polymerization started at this point and it was allowed to continue for 25 minutes at room temperature. Aniline monomer to ammonium peroxydisulfate molar ratio was kept to 1:1.25 [30]. Alternatively, codoped PANi without NaClO was synthesized. Polymerization was carried out in 60 minutes and the HCl/CSA ratios in Table 4 were used. In each case, the resulting dark-green solids were filtered and washed with hydrochloric acid [1 M] and distilled water until the filtrate became colorless. Finally, powders were dried in an oven at 80 °C overnight.
Characterization
UV-Visible spectroscopy
Absorbance spectra from 300 to 900 nm were obtained by a Perkin-Elmer Lambda 35 spectrometer using a set of paired cells of 1 cm of optical path length made of quartz glass. Each sample (0.001 g) was dissolved in 20 mL of distilled water in an ultrasonic bath.
FTIR-ATR (attenuated total reflection) spectroscopy
Transmission FTIR spectra of PANi powder samples were directly examined in an ATR accessory attached to a Perkin-Elmer System 2000 spectrometer. Spectra were recorded in the 4000 to 400 cm-1 region and were corrected for CO2 and H2O content in the optical path.
XRD analysis
Measurements were carried out in a Bruker Focus D8 X-Ray diffractometer using Cu Kα radiation. X-Ray diffraction patterns were recorded from 5° to 70° at the speed of 8°/min.
Scanning electron microscopy
The morphology of the powders was observed using a field emission SEM JEOL JSM-6701F without conductive coating on samples at a 15 kV acceleration voltage.
Conductivity measurements
PANi powders were uniaxially pressed (1.47 MPa) by using a steel die to form pellets of 12.4 mm in diameter. The samples were masked and coated with a Au-Pd coating by using a Denton Vacuum Desk V sputtering system, in order to obtain a centered contact of 5 mm in diameter on both sides. Cooper leads were attached to Au-Pd contact employing silver paste. Resistance measurements were done using a common digital multimeter. Conductivity was determined by standard Van der Pauw two-probe method. A statistical study based on Student's /-distribution with 95% of confidence was conducted to establish the experimental error in the conductivity measurements. This study consisted in the manufacture of 10 samples syn-thetized independently one of the other, and the ratio 0.6/0.4 HCl/CSA was arbitrarily chosen without NaClO, that is, all samples nominally had the same composition. Then, conductivity measurements were carried out and the values statistically treated. The result was σ = 6.8 x 10-2 ± 2.2 × 10-2 S/cm. The same exactly procedure was done on 10 samples with 0.6/0.4 HCl/CSA ratio but in this case using NaClO (cooxidized) and the result was σ = 1.6x10-2 ± 2.8 × 10-3 S/cm.
Acknowledgements
J. E. O. F. is grateful for scholarships granted by CONACyT (229127) and IPN. Authors thank to Mr. Víctor Ramos and Mr. Gerardo González from Metallurgical and Materials Engineering Department - ESIQIE for DRX measurements and SEM micrographs, respectively.
References
1. Ameen, S.; Akhtar, M. S.; Husain, M. Sci. Adv. Mater. 2010, 2, 441-462. [ Links ]
2. Letheby, H. J. Chem. Soc. 1862, 75, 161-163. [ Links ]
3. Diaz, A. F.; Logan, J. A. J. Electroanal. Chem. Interfacial Elec-trochem. 1980, 111, 111-114. [ Links ]
4. Genies, E. M.; Boyle, A.; Lapkowski, M.; Tsintavis, C. Synth. Met. 1990, 36, 139-182. [ Links ]
5. Gaponik, N. P.; Talapin, D. V.; Rogach, A. L. Phys. Chem. Chem. Phys. 1999, 1, 1787-1789. [ Links ]
6. Zhao, C.; Xing. S.; Yu Y.; Zhang W.; Wang, C. Microelectron. J. 2007, 38, 316-320. [ Links ]
7. Liu, Z; Zhou, J; Xue, H.; Shen, L.; Zang, H.; Chen, W. Synth. Met. 2006, 156, 721-723. [ Links ]
8. Kim, J. S.; Sohn, S. O.; Huh, J. S. Sensor Actuator B 2005, 108, 409-413; [ Links ] Hosseini, S. H.; Entezami A. A. Polym. Advan. Technol. 2001, 12, 482-493; [ Links ] Joshi, S. S.; Lokhande, C. D.; Han, S. H. Sensor Actuator B 2007, 123, 240-245; [ Links ] Muthukumar C.; Kesarkar S. D.; Srivastava D. N. J. Electroanal. Chem. 2007, 602, 172-180. [ Links ]
9. Desilvestro, J.; Scheifele W.; Haas, O. J. Electrochem. Soc. 1992, 139, 2727-2736. [ Links ]
10. Qiao Y.; Li C. M.; Bao S. J.; Bao Q. L. J. Power Sources 2007, 170, 79-84. [ Links ]
11. Huang, W. S.; Humphrey, B. D.; MacDiarmid, A. G. J. Chem. Soc. Faraday Trans. 1986, 82, 2385-2400. [ Links ]
12. Shirakawa H. Synth. Met. 2001, 125, 3-10. [ Links ]
13. Gustafsson, G.; Cao, Y.; Tracy, G. M., Klavetter, F.; Colaneri, N.; Heeger, A. J. Nature 1992, 357, 477-479; [ Links ] Bay, R. F. C.; Armes, S. P.; Pickett, C. J.; Ryder, K. S. Polymer 1991, 32, 2456-2460; [ Links ] Samuelson, L. A.; Anagnostopoulos, A.; Alva, K. S.; Kumar, J.; Tripathy, S. K. Macromolecules 1998, 31, 4376-4378; [ Links ] Chevalier, J. W.; Bergeron, J. Y.; Dao, L. H. Macromolecules 1992, 25, 33253331 ; [ Links ] Chen, S. A.; Hwang, G. W. J. Am. Chem. Soc. 1994, 116, 7939-7940. [ Links ]
14. Cao, Y.; Smith, P.; Heeger, A. J. Synth. Met. 1992, 48, 91-97. [ Links ]
15. Cao, Y.; Smith, P. Polymer 1993, 34, 3139-3143. [ Links ]
16. Chen, S. A.; Hwang, G. W. J. Am. Chem. Soc. 1994, 116, 7939-7940. [ Links ]
17. Yin, W.; Ruckenstein, E.; Synth. Met. 2000, 108, 39-46. [ Links ]
18. Ruckenstein, E.; Yin, W.; J. Appl. Polym. Sci. 2001, 79, 80-85. [ Links ]
19. Bhadra, S.; Khastgir, D.; Singha, Nikhil K.: Lee, J. H.; Prog. Polym. Sci. 2009, 34, 783-810. [ Links ]
20. Avlyanov, J. K.; Josefowicz, J. Y.; MacDiarmid, A. G. Synth. Met. 1995, 73, 205-208. [ Links ] Huang, J.; Kaner, R. B. J. Am. Chem. Soc. 2004, 126, 851-855. [ Links ]
21. Wu, C. G.; Bein T. Science 1994, 264, 1757-1759. [ Links ]
22. Chiou N. R.; Lu C.; Guan J.; Lee L. J.; Epstein A. J. Nat. Nano-tech. 2007, 2, 354-357. [ Links ]
23. Virji S.; Huang, J.; Kaner, R. B.; Weiller, B. H. Nano Lett. 2004, 4, 491-496. [ Links ]
24. Baker, C. O.; Shedd, B.; Innis, P. C.; Whitten, P. G.; Spinks, G. M.; Wallace G. G.; Kaner, R. B. Adv. Mater. 2008, 20, 155-158. [ Links ]
25. Sukeerthi S.; Contractor A. Q. Anal. Chem. 1999, 71, 22312236. [ Links ]
26. Huang, J. Pure Appl. Chem. 2006, 78, 15-27. [ Links ]
27. Chien, J. C. W.; Yamashita, Y.; Hirsch, J. A.; Fan, J. L.; Schen, M. A.; Karasz, F. E. Nature 1982, 299, 608-611. [ Links ]
28. Li, X.; Li, X. Mater. Lett. 2007, 61, 2011-2014. [ Links ]
29. Rahy, A.; Sakrout, M.; Manohar, S.; Cho, S. J.; Ferraris, J.; Yang, D. J. Chem. Mater. 2008, 20, 4808-4814. [ Links ]
30. Stejskal, J; Gilbert, R. G. Pure Appl. Chem. 2002, 74, 857867. [ Links ]
31. McCall, R. P.; Ginder, J. M.; Leng, J. M.; Ye, H. J.; Manohar, S. K.; Masters, J. G.; Asturias, G. E.; MacDiarmid, A. G.; Epstein, A. J. Phys. Rev. B 1990, 41, 5202-5213. [ Links ]
32. Yu, H.; Wang, C.; Zhang, J.; Li, H.; Liu, S.; Ran, Y.; Xia, H. Mater. Chem. Phys. 2012, 133, 459-464. [ Links ]
33. Li, G.; Zhang, C.; Li, Y.; Peng, H.; Chen, K. Polymer 2010, 51, 1934-1939. [ Links ]
34. Pan. L. J.; Pu, L.; Shi, Y.; Sun, T.; Zhang, R.; Zheng, Y. D. Adv. Funct. Mater. 2006, 16, 1279-1288. [ Links ]
35. Bhadra, S.; Singha, N. K.; Khastgir, D. Eur. Polym. J. 2008, 44, 1763-1770. [ Links ]
36. Yue, J. ; Wang, Z. H. ; Cromack, K. H. ; Epstein, A. J. ; MacDiarmid, A. G. J. Am. Chem. Soc. 1991, 113, 2665-2671 ; [ Links ] Sinha, S. ; Bhadra, S. ; Khastgir, D. J. Appl. Polym. Sci. 2009, 112, 31353140. [ Links ]
37. Ginder, J. M. ; Epstein, A. J. Phys. Rev. B 1990, 41, 1067410685. [ Links ]
38. Lux, F. Polymer 1994, 35, 2915-2936. [ Links ]
39. Wudl, F. ; Angus, R. O. Jr. ; Lu, F. L. ; Allemand, P. M. ; Vachon, D. J. ; Nowak, M. ; Liu, Z. X., Heeger, A. J. J. Am. Chem. Soc. 1987, 109, 3677-3684. [ Links ]
40. Tran, H. D. ; D'Arcy, J. M. ; Wang, Y. ; Beltramo, P. J. ; Strong, V. A. ; Kaner, R. B. J. Mater. Chem. 2011, 21, 3534-3550. [ Links ]
41. Stejskal, J.; Trchovä, M. Pure Appl. Chem. 2011, 83, 1803-1817. [ Links ]
42. Kuo, C.-T.; Chen, C.-H. Synth. Met. 1999, 99, 163-167. [ Links ]
43. Moon, Y. B.; Cao, Y.; Smith, P.; Heeger, A. J. Polymer Commun. 1989, 30, 196-199. [ Links ]
44. Klug, H. P.; Alexander, L. E. X-Ray Diffraction Procedures. Wiley-Interscience, Ed. New York, 1974. [ Links ]
45. Bhadra, S.; Singha N. K.; Khastgir, D. Polym. Int. 2007, 56, 919927. [ Links ]
46. Pouget, J. P.; Jözefowicz, M. E.; Epstein, A. J., Tang, X.; MacDiarmid, A. G. Macromolecules 1991, 24, 779-789. [ Links ]
47. Lee, K.; Cho, S.; Park, S. H.; Heeger, A. J.; Lee, C.-H.; Lee, S.-H. Nature 2006, 441, 65-68. [ Links ]
48. Laska, J.; Widlarz, J. Polymer 2005, 46, 1485-1495 [ Links ]
49. Xing, S.; Zheng, H.; Zhao, G. Synth. Met. 2008, 158, 59-63. [ Links ]
50. Rahy, A.; Yang, D. J. Mater. Lett. 2008, 62, 4311-4314. [ Links ]
51. Stejskal, J.; Trchovä, M. Polym. Int. 2012, 61, 240-251. [ Links ]
52. Gizdavic-Nikolaidis, M. R., Bennett J., Zujovic Z., Swift S., Bowmaker G. A., Synth. Met. 2012, 162, 1114-1119. [ Links ]
53. Chen S. A., Lee H. T., Macromolecules 1993, 26, 3254-3261. [ Links ]

