










Servicios Personalizados
Revista
Articulo
 Inglés (pdf)
Inglés (pdf)
 Artículo en XML
Artículo en XML Referencias del artículo
Referencias del artículo
 Enviar artículo por email
Enviar artículo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO -
 Accesos
Accesos
Links relacionados
-
 Similares en
SciELO
Similares en
SciELO
Compartir

 Permalink
PermalinkJournal of the Mexican Chemical Society
versión impresa ISSN 1870-249X
J. Mex. Chem. Soc vol.57 no.3 Ciudad de México jul./sep. 2013
Article
Metal-Based Drug-DNA Interactions
Juan Carlos García-Ramos1, Rodrigo Galindo-Murillo2, Fernando Cortés-Guzmán3,4, and Lena Ruiz-Azuara*1
1 Departamento de Química Inorgánica y Nuclear, Facultad de Química, Universidad Nacional Autónoma de México. Avenida Universidad 3000, 04510, México City, México.
2 Department of Medicinal Chemistry, College of Pharmacy, University of Utah, 2000 East 30 South Skaggs 201, Salt Lake City, UT 84112, USA.
3 Instituto de Química, Universidad Nacional Autónoma de México, México DF 04510, México. fercor@unam.mx
4 Centro Conjunto de Investigación en Química Sustentable UAEMex-UNAM, carretera Toluca-Atlacomulco km 14.5, Toluca, México, 50200.
Received June 20, 2013
Accepted July 22, 2013
Abstract
Coordination compounds that bind to DNA have been an active area of research since the discovery of cisplatin and the platinum based drugs. In this review we offer a general overview about transition-metal compounds that binds DNA through several ways and some examples are given to understand the role played by the different factors that promotes it, such as the intercalant ligand and the nature and position of the substituent over it. Several techniques to follow metal-based drugs interactions with DNA are mentioned as well as a brief description of computational techniques that can be used as a powerful tool in order to reach a deep knowledge of the parameters involved in the stabilization of coordination compound-DNA adduct.
Keywords: Coordination compounds, transition metals, DNA-interaction, antitumoral, experimental methods, theoretical methods.
Resumen
El estudio de los compuestos de coordinación que se unen directamente al ADN ha sido un área activa de investigación desde el descubrimiento del cisplatino y compuestos relacionados con éste. En esta revisión se ofrece una visión general sobre los compuestos de metales de transición que se une el ADN a través de varias formas y damos algunos ejemplos para comprender el papel de los diferentes factores que lo promueven, como el ligante intercalante así como de la naturaleza y la posición del sustituyente sobre el mismo. Varias técnicas para seguir las interacciones compuesto de coordinación-ADN son mencionadas, así como una breve descripción de las técnicas computacionales que se pueden utilizar como una herramienta poderosa con el fin de llegar a un conocimiento profundo de los parámetros involucrados en la estabilización del aducto compuesto de coordinación-ADN.
Palabras clave: Compuestos de coordinación, metales de transición, interacción con ADN, anticáncer, métodos experimentales, métodos teóricos.
Introduction
DNA is the library of life and all the knowledge encrypted therein becomes more accessible every day. It has been 10 years now since the Human Genome Project announced that the total human genome was decoded providing us a huge quantity of new specific therapeutic targets [1]. The knowledge of specific targets in rational design of chemotherapeutics is a fundamental factor, principally, for the design of molecules that can be used in the treatment of oncologic diseases. Since the development of cisplatin, DNA became one of the main biological targets for the antitumor compounds [2, 3], even though some researchers consider that this strategy seems unproductive today for the area of new drug development where the knowledge on cancer genomics is suggesting the use of targets selectively expressed, or over-expressed by cancer cells [4]. The intention of this review is to provide several examples of coordination compounds that possess an intercalating ligand in its coordination sphere in order to understand the influence of several factors such as the preferential geometry of the metal center, the nature of the intercalating ligand and the nature, number and position of the substituents over the intercalating ligand in the capacity and selectivity of the coordination compounds to interact with DNA. Also, experimental and theoretical approaches to evaluate the metal-based drugs interactions with DNA are discussed.
DNA structure
A simple unit of DNA known as "nucleotide" is constituted by a five-membered carbohydrate deoxyribose bonded in its C1' to a heterocyclic base that can be either adenine (A), guanine (G), cytosine (C) or thymine (T), and on the other side of the carbohydrate ring, at C5'. A phosphodiester linker bonds the 5'-end of one sugar and the 3'-end of the other. Accordingly, the polymerization of nucleotides gives rise to long, single-stranded polyanionic chains with a well-defined directionality (traditionally described in the 5'- to 3'- direction) which essentially constitutes the primary structure of the nucleic acid [5].
The first DNA structure was published in 1953 by James Watson and Francis Crick [6] without the appropriate recognition to the Rosalind Franklin contributions. Franklin was the first to discover that DNA fibers yield two different diffraction patterns: the "crystalline" or A-form and the "wet" or B-form. John Randall gave Franklin the A-form and Maurice Wilkins the B-form, assigning them each the task of elucidating their molecular structure. However, it was a titanic job due that the samples contain both structures and provided "muddy" diffraction patterns that were near impossible to interpret. After discovering the existence of the A and B forms of DNA, Rosalind Franklin also succeeded in developing an ingenious and laborious method to separate the two forms, providing the first DNA crystals pure enough to yield interpretable diffraction patterns. She then went on to obtain excellent X-ray diffraction patterns of crystalline B-form DNA and, using a combination of crystallographic theory and chemical reasoning, discovered important basic facts about its structure. She discovered that the sugar-phosphate backbone of DNA lies on the outside of the molecule, not the inside as was previously thought. She also discovered that the helical structure of DNA has two strands, not three as proposed in competing theories. She gave quantitative details about the shape and size of the double helix, but the missing piece of the puzzle, how the base paired on the inside of the helix, and thus the secret of heredity itself, was the part provided by Watson and Crick to complete the famously structural model [7]. DNA adopts a double-stranded arrangement as their secondary structure. The two strands of the duplex are held together by hydrogen bonding between bases on opposite strands: the most common "base-pairing" scheme (often called "Watson-Crick base pairing") involves G-C and A-T purine-pyrimidine inter-strand pairs. The overall stability of the double-helical arrangement is further supported by π-π interactions between the stacked hydrophobic aromatic rings of adjacent bases on the polynucleotide chains. This structure forms two distinct grooves labeled major and minor, the major groove is narrow and deep, while the minor groove is wide and shallow [8, 9].
Conformations
DNA is able to adopt a variety of different conformations that are influenced by environmental factors (hydration and ionic strength) and the primary structure (base sequence) of the polynucleotide [10] and are classified as A-, B-, C-, D- and Z-form [11]. However, under physiological conditions, the B-form of DNA is predominant. A-form is a right-handed helix that consists of 11 base residues per full turn with a closer distance (2.25 Å) between the bases and a helix diameter of 23 Å. A-DNA is also characterized by a 20° tilting of the base pairs and their net migration away from the central axis. This produces a more compacted structure with a hollow core. On the other hand, the base pairs of the right-handed B-DNA are stacked 3.4 Å apart with ten and a half base pairs residues per full rotation of the helix and a helix diameter of 20 Å. The minor groove in this structure is 4.8 Å while the major groove is 10.5 Å wide (Figure 1).
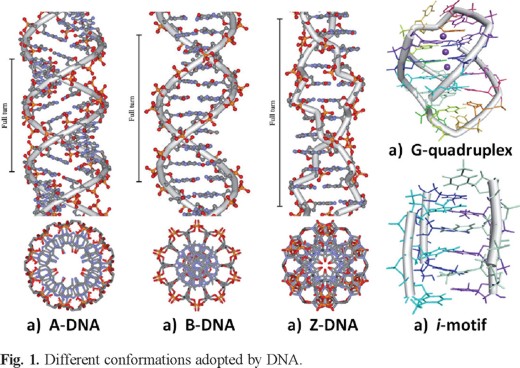
A third (C-DNA) and fourth (D-DNA) allomorphs, both B-like in structure but with reduced rise per residue (3.31 and 3.05 A, respectively) and increased helical twists (38.6-40.0 and 45.0°, respectively), are obtained with Li and Na ions under low hydration conditions, which is when the salt content is between the A- or B-DNA states. Alternating purine and pyrimidine sequences also produces D-DNA form [11].
The Z-DNA conformation is adopted at high salt concentration and is characterized by the adoption of left-handed geometry. The helix in this form is elongated and slender (diameter of 18 Å) and can be induced by the covalent bindings of various metal complexes. It has twelve base pairs per turn with the sugar phosphate adopting a zigzag shape. The orientation of the carbohydrates groups alternates every second unit, revealing a departure from the standard mononucleotide repeating unit of B-DNA in favor of a dinucleotide motif. The minor groove in this arrangement is deeper than that observed in the B-DNA extending all the way down to the axis of the molecule, thus, the major groove is absent here (Figure 1).
DNA can also adopt other conformations such as hairpin, triplex (H-DNA), cruciform, or tetraplex (G-quadruplex and i-motif). In order to form those structures, DNA strands should be folded in a different manner from B-DNA or make unusual base pairs such as Hoogsteen base pairs that are the unusual pairs of hydrogen bonding among nucleic bases compared with Watson-Crick base pairs. Self-complementary sections of single strand DNA can form hairpin structures or cruciform structures, where two opposite hairpins are interacting [12]. Hairpin structures have been identified in genomic DNA and are considered potential sites for controlling gene expression. So, drugs that can target hairpin and cruciform structures in promoter regions may be able to modify transcription and control gene expression [13-16]. Guanine (G)-rich sequences can form G-quadruplex structures consisting of π-π stacking of planar G-tetrads, cyclically bound to each other through eight hydrogen bonds according to the Hoogsteen base pairs [17-19]. G-rich sequences are observed frequently in the promoter region of oncogene and human telomeric DNA [20-22]. The i-motif structure is formed from a cytosine (C)-rich strand at slightly acidic pH or even neutral pH [23]. It is known that C-rich sequences are present in or near the regulatory regions of >40% of all genes, especially in the promoter region of oncogene and human telomeric DNA [24]. The i-motif structure consists of two parallel-stranded C: C+ hemi protonated base-paired duplexes that are intercalated in an antiparallel manner. The i-motif structures are significantly affected by the number of cytosine bases [25], loop length [26], environmental condition [27, 28], and attached or interacting material with the DNA strands [28-30].
Double stranded DNA can also bind to a third strand under acidic conditions to form a triple helix, also known as H-DNA [31-33]. The bases of the third strand bind to the existing base pairs via Hoogsten base pairing, where T binds to A in a novel fashion and protonated C binds to G, forming the triplets T•AT and C•GC complementary sequence. Triple helices are not as stable as duplex, and are thought to relieve torsional stress caused by supercoiling.
Modes of binding of bioinorganic structures to DNA
A search using the Nucleic Acids database [34] produces 247 crystal and NMR structures of DNA with various types of binders, from which only a handful relates to any kind of bioinorganic or metal-containing drug. Some examples of experimental drug-DNA interactions using transition metal compounds include Ru [35], Co [36], Ni, Cu, Zn [36], and rare-earths compounds [37]. Since DNA offers many potential binding sites due to its size and complexity, coordination compounds are able to bind to DNA via two main interaction modes: irreversible (covalent or coordination binding) and reversible (intermolecular association). The latter binding mode can be further divided into electrostatic interactions, grove binding and intercalation.
Covalent binding
Coordination compounds may exhibit a preference for a particular binding mode or nucleotide sequence depending on the size and shape of the molecule. Cisplatin is a well-known antitumor drug which principal target is DNA. It is responsible for the cure of over 90% of testicular cancer [38] and plays an important role in the treatment of other kinds of cancer such as ovarian, bladder, head and neck, lymphomas and melanomas [39]. Even though the great knowledge achieved, cisplatin is only effective in a limited range of cancer and also produce severe side-effects: often causing severe nausea and vomiting, bone marrow suppression and kidney toxicity. In spite of that, cisplatin is still one of the world's best-selling anticancer drugs.
The action mechanism of cisplatin has been extensively studied as well as the solvation process that it suffers to produce the "activated" molecules cis-[PtCl(H2O)(NH3)2]+ and cis-[Pt(H2O)2(NH3)2]2+ [39-46]. Antitumor activity of solvated cisplatin derives from its capability to form bifunctional DNA cross-links [47]. The predominant adducts formed by cisplatin with DNA are guanine-guanine (GG) intrastrand cross-links throughout the coordination of Pt to N7 of guanine (Figure 2). The coordination to N7 of guanines is not surprising as nitrogen N7 atoms of purines are the most electron-dense and accessible sites in DNA for electrophilic attack by platinum. They are exposed in the major groove of the double helix, and not involved in base pair hydrogen-bonding. This particular platinum-induced kink in the DNA is considered to be the critical lesion due to the chain of events that the biomolecular recognition eventually provokes in the cell [47-51].
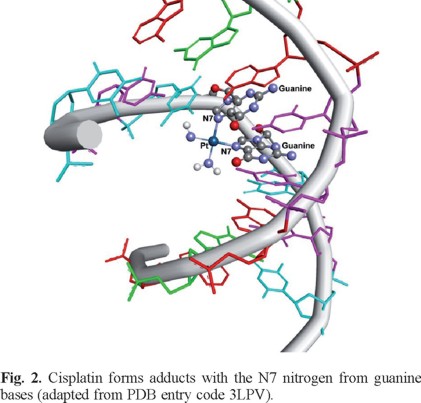
The significant prevalence of intrastrand GG cross-links over any other adduct has been examined in the context of contributions of H-bonding, the electrostatics in the vicinity of the binding site, and the steric effects of adduct conformation. It has been argued that hydrogen bonding between the NH3 of the DNA-cisplatin complex and O6 of a nearby guanine—not possible for adenine—has a large impact on the adduct configuration, being essential for stability and eventual cytotoxic activity [52-54].
Some other platinum derivatives present a covalent interaction with DNA. Platinum (IV) complexes with octahedral geometry are believed to be reduced to platinum (II) in vivo [55]. Satraplatin ((OC-6-43)-bis(acetato)amminedichloro(cyclohexylamine) platinum, JM216) is the most successful of the platinum(IV) complexes [56], on the other hand, iproplatin (cis-dichloro-transdihydroxy-bis-isopropylamine platinum(IV)) and tetraplatin (1,2-diaminocyclohexane tetrachloroplatinum (IV)) [57] are also undergoing clinical development. A number of multinuclear platinum complexes have also been developed [58, 59], some of which have shown activity in both cisplatin-sensitive and cisplatin resistant cell lines.
All mononuclear platinum complexes could form intrastrand and interstrand adducts with DNA. When interstrand lesion is formed, massive distortions of the B-DNA are observed. Similarly, the intrastrand lesion, while it forms much more readily than the interstrand lesion, it induces mutational events via the distortion of its nucleic acid target [60]. These detrimental issues have been addressed by a generation of dinuclear Pt complexes which possess two Pt (II) centers, each of which carries only one chloride ligand thus relegating each Pt (II) capable of only one coordination event. Farrell and his group designed and synthesized binuclear platinum (II) complexes and studied their interactions with calf thymus DNA and a small 49 base pair oligodeoxyribonucleotide, see Figure 2 [61]. Owing to the presence of the pyridyl ligands, this compound induces a much higher degree of DNA unwinding than that seen with either of the ammonia-bound complexes, as well as the mononuclear trans-[PtCl2-(py)2]. Similarly, to the mononuclear compound [(Pt(trans-(py)2Cl)2-µ-(diamminobutane)]2+ stabilizes B-form DNA while the other complexes with amino groups instead pyridine rings induce B-Z transitions upon DNA binding. These alterations likely involve the ability of [(Pt(trans-(py)2Cl)2-µ-(diamminobutane)]2+ to undergo π-stacking interactions upon DNA association which in turn, disfavors the Z-DNA conformation. Importantly, interstrand-cross links formation is very efficient for all three complexes with yields of 80%, 52%, and 41% for [(Pt(cis-(NH3)2Cl)2-µ-(diamminobutane)]2+, [(Pt(trans-(NH3)2Cl)2-µ-(diamminobutane)]2+, and [(Pt(trans-(py)2Cl)2-µ-(diamminobutane)]2+ respectively. Further, only Further, only [(Pt(trans-(NH3)2Cl)2-μ-(diamminobutane)]2+ was capable of intrastrand adduction.
DNA binding of BBR3464 was the formation of {Pt,Pt} inter-strand cross-links (IXL), expected to be structurally distinct from those formed by cisplatin because each Pt unit will bind to one strand of the DNA duplex [62]. For this compound the crosslinks occur not only in the ''normal'' 5'-5' direction, since the DNA helix is normally read from the 5'-side, but also in the ''opposite'' antiparallel 3'-3'direction [63]. The directionality is dependent on the nature of the cross-link. Interestingly and according to Farrell, this is the unique example of anti-cancer drugs behaving in this manner [64, 65].
Trinuclear platinum complex BBR3464 is one of the most studied of this kind [66-69]. This compound has shown a higher potency than cisplatin in vitro [70-72], while in vivo studies have shown it to be effective towards human xenograft tumors with intrinsic or acquired resistance to cisplatin [73]. In phase II clinical trials only partial responses have been observed with cisplatinresistant ovarian and non-small cell lung cancers [74]. Even though Phase I and II clinical studies gave preliminary indications of activity in melanoma, pancreatic, lung and ovarian cancers, further phase II studies have shown to have minimal antitumor activity against gastric carcinomas, with significant hematological toxicities encountered by patients, the authors conclude that further studies with BBR3464 in gastric or gastro-esophageal adenocarcinoma are not recommended [75].
Groove binding
Molecules normally reach DNA through one of the grooves and react to either the backbone or the nucleobases. Reversible intermolecular associations such as electrostatic binding occur due the interaction between cations with the negatively charged phosphate backbone at the exterior surface of the DNA helix [76]. Some of the most common ligands which have been studied as intercalator are shown in Fig. 3.
Major groove
Ruthenium (II) polypyridyl complexes can bind by direct interactions in either the major or minor groove of DNA due to their great diversity in overall shape, size, hydrogen-bonding potential, polarizability of ligands and charge. Even though if the structure has an adequate intercalation motif, compounds with bulky substituent preferentially occupy the major groove. Examples of this kind of complexes include the ruthenium derivatives with the ability to hydrogen-bond that appear to favor major-groove binding like Λ-[Ru(TMP)3]2+ (TMP = 3,4,7,8-tetramethyl-1,10-phenanthroline) [77] and [Ru(phen)2(11-(9-acridinyl)dipyrido[ 3,2-a:20,30-c]phenazine)]2+ which present a preference to intercalate on GC rich region of the major groove than the AT rich one [78]. Copper compounds also present this binding mode, like [Cu(ferrocenylmethylbis(2-pyridylmethylamine)(dppz)](ClO4)2 which shows dual chemical nuclease activity involving both metal centers and displays efficient photo-induced DNA cleavage activity in visible laser light of 458 and 568 nm and also in red light of 647 nm within the photodynamic therapy (PDT) window [79] or [Cu(salicylaldehyde-N(4)-phenylthiosemicarbazone) (dppz)] which also present nuclease activity [79].
Tetracationic triple helical supramolecular cylinder [Fe2(C25H20N4)3]Cl4 was one of the first reported examples of this type of complexes that interact with DNA major groove. Major groove interaction was suggested by the DNA kinking observed in atomic force microscopy (AFM) images of the interaction between the helicate and plasmidic DNA pBR322. The major groove binding was confirmed by NOESY experiments [80]. Further studies of the interaction of this helicate structure with DNA show that exist an enantiomeric preference for the binding process that is reflected in the DNA deformation degree [81]. Binding strength and groove selectivity can be modified adding methyl groups to the helicate that produce width increase or lengthening of the structure. Increasing the width reduces the DNA binding strength, the bending and coiling effect and the groove selectivity of the enantiomers compared with the parent compound, while the lengthening results in DNA interactions similar to those of the parent compounds, with an increased preference of the P enantiomer for the minor groove indicating an enhancement of mode (P enantiomer of these iron supramolecular helycates deviates polarized light to the left) [82]. Another examples of helicate structures that provide certain groove selectivity degree are the enantiomerically pure dinuclear complexes of the form [Ru2(1,10-phen)4L1]4+ and [Ru2(bpy)4L1]4+, where L1 = (C5H4N)C=N(C6H4))2CH2). In contrast to the di-iron helicates, the phen complexes show DNA sequence effects with Δ, Δ-[Ru2(phen)4L1]4+ binding preferentially to GC and Λ, Λ-[Ru2(phen)4L1]4+ to AT [83].
Intercalation
Intercalation is distinguished by the insertion of generally planar polycyclic aromatic systems between the stacked base pairs of DNA. This interaction is stabilized by the overlap of the π-clouds of the intercalator and the nearby nucleobases [84] and can occur from either the major [85] or the minor groove [86]. Intercalation increases the separation of adjacent base pairs, and the resulting helix distortion is compensated by adjustments in the sugar-phosphate backbone and, generally, by an unwinding of the duplex. These systems have a planar surface area of approximately 28 Å2 [87] and they must be positively charged so that electrostatic interactions can occur between the intercalating molecule and the negatively charged DNA [88]. The inclusion of a metal center into the design of intercalating compounds gives rise to a greater range of geometric diversity and structural flexibility than any purely organic molecules can achieve.
Due the remarkable results obtained with cisplatin, for much time was believed that compounds with square geometries possessing an intercalator moiety were the perfect candidates for the accomplishment of the interaction with DNA. In 1974, the intercalative binding of platinum (II) complexes containing the bidentate ligands bpy, phen and terpy were investigated using X-ray diffraction and electrophoresis [59, 89, 90]. However, intercalation is not restricted to square planar complexes, a series of chiral octahedral complexes using zinc [91], iridium [92], iron [93, 94], cobalt [95, 96], osmium [97, 98], rhodium [99-101] and ruthenium [75-105, 123, 124, 141-148] as well as tetrahedral complexes such as [Co(phen)2]2+ [102], [Cu(phen)2]2+ [103], were synthesized and their interactions with DNA investigated.
A series of related platinum (II) compounds containing bpy, phen and TMP as intercalating ligand and ethylendiamine (en) or diaminopropane completing the coordination sphere were tested against the L1210 murine leukaemia cell line to assess their cytotoxicity.
The effect of the intercalating moiety in metallodrugs was investigated by comparing the cytotoxicity of different methyl-substitution on the phenanthroline of [Pt(R-phen)(en)]2+ complexes [104], where R-phen = phen, 4-Me-phen, 5-Me-phen, 4,7Me2-phen, 5,6-Me2- phen and TMP in the L1210 cell line. These achiral complexes present IC50 values in the range 0.7-2 mM that suggest to the investigation group that the combination of intercalating and ancillary ligand coordinated to the platinum has a significant effect on the biological activity of these complexes [105]. These experiments also did show that methyl substitution on specific endocyclic carbon atoms of phen can affect the biological activity of a complex. The order of activity is 5,6-Me2-phen > 5-Me-phen > phen > 4-Me-phen = 4,7-Me2-phen = TMP. NMR studies revealed that they all insert into the hexanucleotide d(GTCGAC)2 from the minor groove, predominantly between the T2A5 and C3G4 base pairs. The NMR evidence also indicated that the intercalator is inserted into the DNA with the ethylendiamine moiety in the minor groove. It is probable that the observed enhancement in biological activity is due to favorable hydrogen bonding or steric interactions. For the complexes [Pt(4Me-phen)(en)]2+, [Pt(4,7-Me2-phen)(en)]2+ and [Pt (TMP)(en)]2+, results from DNA-NMR binding experiments suggest that the hydrogens of the methyl groups on C4 and C7 are close to the base pairs and possibly interacting unfavorably and this may result in the reduced biological activity of these complexes. In addition, it is possible that the methyl groups interact with phosphodiester groups of the sugar-phosphate backbone, affecting the depth of intercalation [104]. Most platinum (II) intercalators binding have association constants in the range of 104-107 M-1. Variance in binding strength appears to be due to differences in the charge and extent of the aromatic system of individual molecules, with the general trend being phen > terpy > bpy [106-108].
Similar results were reported by Chikira [109] employing ternary Cu(II) complexes with phenanthroline and amino acids, [Cu(phen)(α-aminoacidate)], shown that the three-fused aromatic rings in phenanthroline are critical for the intercalative binding of the complexes. The intercalative binding was promoted by 5,6-dimethyl groups on the phenanthroline ring, whereas it was disturbed by 2,9-dimethyl groups, indicating that the planarity of the coordination sphere is important for the intercalative binding. For these compounds, the inhibitory concentrations are in the micromolar range in several cell lines such as cervix (HeLa and SiHa), breast (MCF-7), and colon (HCT-15). Even though these ternary Cu(II) coordination compounds have shown a remarkable cytotoxic, cytostatic, genotoxic and antitumoral activity [110], the paramagnetism of these molecules made hard the study of interaction with oligonucleotides, studies that were done as a routine analysis with metals such as Pt(II) and Ru(II) employing NMR spectroscopy.
This is the case of mononuclear ruthenium (II) complexes that were extensively studied in the middle of the 1980s and during the 1990s [85, 111-140]. One of the first examples of transition metal complexes that interact with DNA extensively studied was Λ- and Δ-[Ru(phen)3]2+. Different biological activity was established for both isomers by studies carried out on mice [141]. The results of these experiments allowed setting out the hypotheses that the interaction of these complexes with the biological system was physical instead chemical, and that the biological effect must be due to the overall three-dimensional shape of the complex, rather than simply the constituent ligands (Figure 4). Association constants are reported to be of the order of 105-106 M-1 [102].

Barton [142-144] suggested that binding of [Ru(phen)3]2+ could occur by two possible modes: intercalation and surface interaction. However, other studies published some years later employing linear and circular dichroism studies suggested that both isomers do not intercalate [144]. DNA interaction discrepancies were solved by posterior NMR studies where were established that [Ru(phen)3]2+ exhibit different binding modes, depending on the chirality of the enantiomer of the complex, the DNA sequence and, for the Δ-enantiomer, the concentration of the complex, at high metal complex to DNA ratios, the minor groove is saturated by surface bound complexes, whereas the major groove is partly filled in a partially intercalative geometry [137, 139, 145].
On the other hand, bis-intercalating compounds consisting of two intercalating moieties joined by a linking chain or spacer were developed to improve cytotoxicity. Bis intercalating compounds may have higher affinities due to their increased charge and ability to span a greater number of DNA base pairs unobtainable by mono intercalators [146]. For platinum derivatives of this kind of compounds the cytotoxicity was reported to depend on three variables; linker length, charge density and counter ions [147]. Complexes with shorter linkers displayed higher activity than those with longer linkers [147]. The more highly charged complexes were also more cytotoxic [146, 147]. The high charge density and greater electrostatic stress of the shorter linked complexes is believed to increase the platination of DNA, which leads to their greater cytotoxicity [147].
Dimers of rhodium (III) complexes such as [Rh(bpy)2(phi)]3+ (phi = 9,10-phenanthrenequinone diimine) and[Rh(phi)2(bpy)]3+ have also been found to show some DNA recognition. Dimers of Δ-[Rh(4,4'-diphenylbpy)2(phi)]3+ (4,4'-diphenylbpy = 4,4'-diphenyl-2,2'- bipyridine) specifically targeted the palindromic DNA sequence 5'-CTCTAGAG- 3' [122, 148]. These rhodium (III) complexes demonstrated the significance of shape complementarity for specific binding between the octahedral complexes and DNA. Complexes with sterically bulky methyl or phenyl groups on the non-intercalating ligands showed sequence preferences [149].
As well as in platinum complexes, Rh(III) derivatives combining intercalating and coordinated easily leaving groups have also been studied [150-151]. 2D NMR shows combined coordinative and intercalative interactions between the dirhodium(II) complex cis-[Rh2(dap)(μ-O2CCH3)2(η1-O2CCH3)(CH3OH)] (O2CCH3)14 (dap = 1,12-diazaperylene) and the duplex (5'-GGAAGTTGAGAG-3')/(5'-CTCTCAACTTCC-3'). One rhodium atom binds coordinatively to the N7 of A6 base, while the dap ligand intercalates classically into the central A6pA7 base step.
Besides NMR spectroscopy and X-ray diffraction, the atomic force microscopy has been employed in the study of metallodrugs-DNA interactions in recent years with remarkable results. In this field the indispensable reference is the group of Professor Virtudes Moreno, who has studied the interaction of an endless number of coordination compounds. Among the studied compounds some of them present antitumor activity such as derivatives of Pt (II) and Pd (II) [152-154], Ru (II) [155-157], Co (III) [158], Cu (I) [159], Cu (II) [160]. This technique made possible to identify the interaction of paramagnetic compounds such as Casiopeínas (Cas III-ia, [Cu(4,4'-dimethyl-2,2'-bipyridine)(acetylacetonate)]NO3; Cas III-Ea, [Cu(4,7-dimethyl-1,10-phenanthroline)(acetylac etonate)] NO3, and Cas II-gly, [Cu(4,7-dimethyl-1,10-phenanthroline)(glycinate)]NO3) with plasmidic DNA pBR322 establishing interaction differences derived from the structural modifications [160].
In addition to the antitumor compounds, some metallodrugs have been designed to be used in the treatment of other diseases also trying to reach DNA as main target. Some examples are oxovanadium compounds tested as antileishmanial [161] and antitrypanosomal [162, 163] agents. Metals usually employed to get antitumor agents such as Pt(II), Pd(II) were recently tested against Trypanosoma cruzi with very good results [164]. Ru(II) compounds with phosphine and diimine ligands were tested against tuberculosis [165], while Au(III) compound [Au(dppz)2]Cl3, [Cu(dppz)2]BF4 and trans-platinum (II) compounds with chloroquine have shown noteworthy results in the proliferative inhibition of Leishmania mexicana cultures [166-168].
Although a lot of experimental work has been done studying the interaction of coordination compounds with DNA, much of this knowledge is still the peak of the iceberg, due that in many cases the information obtained with experimental designs that do not involve expensive procedures and equipments is extremely poor. However, in recent years the computational methods have been suffering an incredible growth related with the size of the systems that can be calculated and the time expended to get results. The next part of this review is focused in the description of some computational approaches employed in the investigation of metal based drugs-DNA interactions.
Computational Chemistry approach of coordination compounds binding to DNA
While extensive research has been done about drug-DNA binders, work about coordination compounds interacting with DNA is still an exciting and active field of research. Experimental techniques and analysis used for routine DNA studies can be extrapolated to coordination complexes in most cases and this is also true with Computational Chemistry tools, except by the fact that transition metals are still complex to simulate due to their multiple oxidation states and resulting geometry change, therefore, special care has to be accounted when generating force field parameters or applying a Quantum Mechanical method to these molecules. Extensive work has been done to understand the interaction of metals and transition-metals with DNA as cations present in the solvent (for an in-depth review, refer to the work by Sponer and co-workers [169]). In this manuscript, we focus on research about metal-containing compounds that interact directly with DNA.
Molecular Dynamics studies
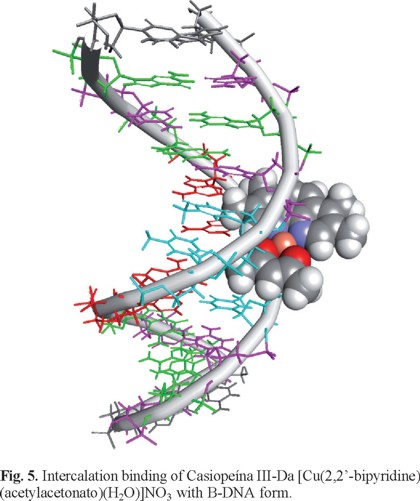
Studies using Molecular Dynamics (MD) are numerous and provide insight about the general structure of the mechanism of binding. Even if no electronic effect is taken into account, these simulations provide useful information of the dynamics not only in the place where the metal-compound is binding but also the conformational changes of the whole system. The AMBER and the CHARMM family of force fields for nucleic acids are able to simulate a stable B-DNA chain in the micro second realm and realistic sampling of the global and local parameters is now possible [170]. MD also allows refinement based on Nuclear Overhauser Effect (NOE) restraints of distance, angle and penalty functions based on NMR spectra. This type of analysis tools is probably the most efficient way to study the structure of a metal compound and the way it binds to nucleic acids, but it requires the restrain information obtained by NMR spectroscopy. The technique has been used to build models and study the structure of many systems [171-173] and also in depth study of DNA-protein binding and triple-helix DNA [174]. In the field of metal-DNA binding, recent work includes studies of the ruthenium complex [Ru2bidppz(bipy)4]4+ and the dodecamer [d(CGCGAATTCGCG)]2 using NMR spectroscopy showed a minor-groove binding interaction that was stable after 10ns of simulation (Figure 4) [175]. Force Field (FF) parameters of small organic molecules are available and provide acceptable results, however, these general force fields do not have the parameters to describe a bioinorganic compound which includes a transition metal, so this is a concern that has to be resolved to be able to run a MD simulation. There are several options available that will generate the bonded and non-bonded parameters around a metal center, one of this is the Metal Center Parameter Builder [176, 177] available in the Amber programs [178]. If the X-ray or NMR structure of the metal-compound is available, then it will be relatively easier to generate the required FF parameters using the bond, angle and dihedral values from the experimental structure. Non-bonded parameters are available for a number of metals [179]. The resulting FF values can be used to recreate the metal-compound and run MD for several nanoseconds to guarantee its stability and correct representation for the simulation. An example of this methodology is shown in our previous work using the copper-compound [Cu(2,2'-bipyridine)(acetylacetonate)(H2O)]+ (Casiopeína®, [180], Since an X-ray of the binding of this copper compounds is still not available, we manually docked the molecule in 4 possible starting binding configurations with the B-DNA and run MD for 100ns to study the energetics of each resulting trajectory (Fig. 5) [181].
If no experimental structure is available, Quantum Mechanical (QM) methods can be used to optimize an initial structure and with this structure, generate the appropriate FF parameters. In a recent work from Liu, Chen and Tang with a new family of mononuclear copper and dinuclear copper-platinum nucleases Density Functional Theory (DFT) methodology was used to generate the optimized geometries. FF parameters for the nucleases, minus the copper and platinum atoms, were obtained from the Generalized Amber Force Field (GAFF) using the programs available in AMBER11. The charge calculation for each atom was obtained following the Restrained Electrostatic Potential (RESP) procedure that allows a multi-conformation, multi-orientation approach, yielding a more accurate representation of the electronic distribution of the molecule [182]. As mentioned above, van-der-Waals and Lennard-Jones parameters for both metals where adopted from available databases and previous work. B-DNA was generated using the nucgen program available in AMBER11. To generate the initial structure prior to the MD simulation, docking methodology was used over 1000 docking predictions using Autodock4.0. Docking methodology will sample through different conformations between two molecules evaluating the total interaction energy for every conformation until a certain threshold is reached. In this case, the metal binders where located among the minor groove of the B-DNA chain. With this starting structure, MD was run for 40ns. Information about the structural changes and movement of the binders along the groove is extracted from the trajectories and information about the global and local inter and intra base conformation.
Quantum Mechanical studies
The use of Quantum Mechanical methods allow full electronic study of the system and considerable reduction of empirical approximations, although, this methods are extremely computational and memory demanding and the representation of solvent interactions are limited. Explicit solvent representation is simply not reachable with current methods, but one can use a previously generated Molecular Dynamics simulation with explicit solvent and transfer specific water molecules for the QM treatment. It is important to mention that transition metals contain a considerable amount of electrons which will take a lot of computing time. Although is important to include every electron of the metal atom, in most cases the core electrons are not used for reactions or energetics. This can be simulated using effective core potentials (ECP) which "freezes" core electrons and facilitate the electronic representation of the metal, requiring less time to achieve conversion. A simple approach to study these interactions is using isolated bases and nucleotides, basically, stripping apart the DNA or RNA chain. This allows the use of high-level computational methods and analysis procedures. An example is the work done by Hirao and Matsubara using divalent cyclen-coordinated metal complexes (Zn, Cu and Ni) where the metal center is treated with the LANL2dz ECP basis set, the rest of the atoms using the 6-31G∗ and using the B3LYP functional. They study the binding of DNA bases guanine, thymine and uracil to the metal complexes and the effect of the solvent using the polarized-continuum-model (PCM) approximation. They report detailed information about the charge transfer and electrostatic interactions using this methodology [183]. Another advantage about QM methods is the possibility to obtain spectroscopic data (for example: UV-VIS, fluorescence and RAMAN), this allows a way to compare between experiments and simulations starting with specific configurations [184].
Hybrid QM/MM studies
Quantum Mechanics and Molecular Dynamics are both methods that give detailed information about some aspect of a molecular system, each with its own advantages and flaws. In an attempt to bring up together the best of both worlds, Warshel and Levitt [185] created a hybrid approach in which the system was split between two zones or layers. The high layer or QM part is where the reaction is taken place, as it occurs in the active site inside enzymes, and the low layer or MM part, which in this case is the rest of the tertiary structure of the same protein. In most cases, the dynamics and fluctuations of the whole protein are not directly involved in the reaction mechanism; therefore, it is not required to employ such a detailed level of calculation like the ones attainable with a QM method. On the contrary, on the active site, we want to study bond breaking, bond forming, electronic interactions, etc., and if possible avoid the use of empirical approximations. The main idea of the QM/MM approach is to split the system of study so that the binder and the area involved in the binding process are simulated with QM and the rest of the system is modeled with MD. The energy of the system is then given by equation:
Eele ≈ EQM + EMM + EQM/MM
EQM and EMM can be obtained using any of the methodologies already mentioned. The differences between the available QM/MM implementations reside in how to calculate the term EQM/MM. Most common methods include ONIOM [186, 187], Car-Parinello QM/MM [188], effective fragment potential [189] and empirical valence bond method [190].
One of the earliest works using hybrid methods to study the binding of coordination compounds to DNA was done using the cisplatin system with the Car-Parinello/MM scheme [191, 192]. This methodology has been proved efficient to study the non-covalent interactions and dynamics of new cisplatin derivatives with DNA [193]. The ONIOM partitioning scheme was applied in the study of Ruthenium compounds binding to B-DNA. First, the DNA molecule is generated and solvated using explicit solvent and counter ions are added. The system is then heated and equilibrated over a 100ps time frame using MD simulations. The resulting structure is extracted from the solvent and the ruthenium complex is manually placed near two guanine bases. The QM layer was defined as the metal complex, including the two guanine bases, and the MM part is the rest of the DNA chain. This allowed to generate a fully optimized geometry of the placement of the metal complex and the guanine bases and then, use only those three molecules to run a series of transition states to study two possible reaction mechanisms [194].
Even though the information of the rest of the DNA chain is lost, following this methodology allows to study a possible reaction mechanism starting from an explicitly solvated structure.
Final remarks
The development of new coordination compounds that specifically interact with DNA—one of the main targets used in the treatment of several diseases, principally cancer—, is the Holy Grail for all the researchers devoted to medicinal chemistry. The treatment of cancer focusing on the interruption of reproductive cycle has been a main objective for many years. Now, with the knowledge of complete genome and the fast growth experimented by the epigenetic, nucleic acids have to be one of the main targets of which chemists have to aim when developing new compounds. The use of transition metals gives a superb tool to drug chemists to develop and study molecules capable of obtaining specific DNA-drug interactions considering the multiple options of d-block metals available in the periodic table. Transition metals are dynamic in geometry, electron affinity and reactivity, making them excellent choices to feed the ongoing field of antineoplastic drug development. Even though the enormous advantages provided by the employment of transition metals related with the three-dimentional arrangement and with the knowledge of the fundamental role played by the intercalating moieties to provide a reversible interaction with DNA, the fundamental factors of this interactions still posses greatest gaps inasmuch as the results provided by experimental designs that do not involve expensive protocols and equipments are extremely poor to identify the specific interactions due the lower energetic changes involved, making clear the use of methodologies such as computational chemistry to help solve these problems. In this review we try to provide a general scope about experimental and theoretical approaches that are used in current research and some of the limitations and success stories found. We consider this area of research a hot and current topic considering the improved experimental and analysis techniques available to synthesize novel coordination compounds designed to accomplish determined task, such as direct and specific interaction with a particular region of DNA.
Acknowledgements
JCGR acknowledge CONACYT for the scholarship grant. The authors acknowledge CONACyT 179119, ICyT DF PIN-VII-32 and PAPIIT UNAM IN 218013 and IN 208113 for the financial support.
References
1. Finishing the euchromatic sequence of the human genome. Nature 2004, 431, 931-45. [ Links ]
2. Huang, R.; Wallqvist, A.; Covell, D. G. Anticancer metal compounds in NCI's tumor-screening database: putative mode of action. Biochem. Pharmacol. 2005, 69, 1009-39. [ Links ]
3. Pizarro, A. M.; Sadler, P. J. Unusual DNA binding modes for metal anticancer complexes. Biochimie 2009, 91, 1198-211. [ Links ]
4. Sava, G.; Jaouen, G.; Hillard, E. A.; Bergamo, A. Targeted therapy vs. DNA-adduct formation-guided design: thoughts about the future of metal-based anticancer drugs. Dalton Transactions 2012, 41, 8226-34. [ Links ]
5. Alberts, B.; Johnson, A.; Lewis, J.; Raff, M.; Roberts, K.; Walter, P. Molecular Biology of the Cell; 5th ed.; Garland Science, 2002; p. 1268. [ Links ]
6. Watson, J. D.; Crick, F. H. C. A Structure for Deoxyribose Nucleic Acid. Nature 1953, 171, 737-738 (1953). [ Links ]
7. Maddox, B. The Double Helix and the "wronged heroine". Nature 2003, 421, 407-8. [ Links ]
8. Hunter, C. A.; Lawson, K. R.; Perkins, J.; Urch, C. J. Aromatic interactions. J. Chem. Soc., Perkin Transactions 2 2001, 651-669. [ Links ]
9. Sriram, M.; Wang, A. H.-J. Structure of DNA and RNA. In Bioorganic Chemistry: Nucleic Acids; Hecht, S. M., Ed.; Oxford University Press: New York, 1996; pp. 105-143. [ Links ]
10. Shakked, Z.; Guerstein-Guzikevich, G.; Eisenstein, M.; Frolow, F.; Rabinovich, D. The conformation of the DNA double helix in the crystal is dependent on its environment. Nature 1989, 342, 456-60. [ Links ]
11. Neidle, S. Oxford Handbook of Nucleic Acid Structure; Oxford University Press: New York, NY, 1999; p. 8. [ Links ]
12. Wadkins, R. M.; Vladu, B.; Tung, C. S. Actinomycin D binds to metastable hairpins in single-stranded DNA. Biochemistry 1998, 37, 11915-23. [ Links ]
13. Hägerlöf, M.; Papsai, P.; Chow, C. S.; Elmroth, S. K. C. More pronounced salt dependence and higher reactivity for platination of the hairpin r(CGCGUUGUUCGCG) compared with d(CGCGTTGTTCGCG). J. Biol. Inorg. Chem. 2006, 11, 974-90. [ Links ]
14. Brown, D. R.; Kurz, M.; Hsu, V. L.; Kearns, D. R. Formation of multiple complexes between actinomycin D and a DNA hairpin: structural characterization by multinuclear NMR. Biochemistry 1994, 33, 651-664. [ Links ]
15. Henderson, P. T.; Armitage, B.; Schuster, G. B. Selective photocleavage of DNA by anthraquinone derivatives: targeting the single-strand region of hairpin structures. Biochemistry 1998, 37, 2991-3000. [ Links ]
16. François, J.; Thuong, N. T.; Hélène, C. Recognition and cleavage of hairpin structures in nucleic acids by oligodeoxynucleotides. Nucl. Ac. Res. 1994, 22, 3943-3950. [ Links ]
17. Phan, A. T.; Kuryavyi, V.; Patel, D. J. DNA architecture: from G to Z. Curr. Opin. Struct. Biol. 2006, 16, 288-98. [ Links ]
18. Huppert, J. L. Structure, location and interactions of G-quadruplexes. The FEBS Journal 2010, 277, 3452-8. [ Links ]
19. Davis, J. T. G-quartets 40 years later: from 5'-GMP to molecular biology and supramolecular chemistry. Angew. Chem. Int. Ed. 2004, 43, 668-98. [ Links ]
20. Hanahan, D.; Weinberg, R. A. The Hallmarks of Cancer. Cell 2000, 100, 57-70. [ Links ]
21. Brooks, T. A.; Hurley, L. H. The role of supercoiling in transcriptional control of MYC and its importance in molecular therapeutics. Nature Rev. Cancer 2009, 9, 849-61. [ Links ]
22. Brooks, T. A.; Kendrick, S.; Hurley, L. Making sense of G-quadruplex and i-motif functions in oncogene promoters. The FEBS Journal 2010, 277, 3459-69. [ Links ]
23. Guéron, M.; Leroy, J. The i-motif in nucleic acids. Curr. Opin. Struct. Biol. 2000, 10, 326-331. [ Links ]
24. Huppert, J. L.; Balasubramanian, S. G-quadruplexes in promoters throughout the human genome. Nuc. Ac. Res. 2007, 35, 406-13. [ Links ]
25. Mergny, J.; Lacroix, L.; Han, X.; Leroy, J.; Helene, C. Intramolecular Folding of Pyrimidine Oligodeoxynucleotides into an i-DNA Motif. J. Am. Chem. Soc. 1995, 117, 8887-8898. [ Links ]
26. Kendrick, S.; Akiyama, Y.; Hecht, S. M.; Hurley, L. H. The i-motif in the bcl-2 P1 promoter forms an unexpectedly stable structure with a unique 8:5:7 loop folding pattern. J. Am. Chem. Soc, 2009, 131, 17667-76. [ Links ]
27. Zhou, J.; Wei, C.; Jia, G.; Wang, X.; Feng, Z.; Li, C. Formation of i-motif structure at neutral and slightly alkaline pH. Molecular bio-Systems 2010, 6, 580-6. [ Links ]
28. Miyoshi, D.; Karimata, H.; Sugimoto, N. Hydration regulates thermodynamics of G-quadruplex formation under molecular crowding conditions. J. Am. Chem. Soc. 2006, 128, 795-763. [ Links ]
29. Shin, S. R.; Jin, K. S.; Lee, C. K.; Kim, S. I.; Spinks, G. M.; So, I.; Jeon, J.-H.; Kang, T. M.; Mun, J. Y.; Han, S.-S.; Ree, M.; Kim, S. J. Fullerene Attachment Enhances Performance of a DNA Nanomachine. Adv. Mat. 2009, 21, 1907-1910. [ Links ]
30. Li, X.; Peng, Y.; Ren, J.; Qu, X. Carboxyl-modified single-walled carbon nanotubes selectively induce human telomeric i-motif formation. Proc. Nat. Ac. Sc. USA 2006, 103, 19658-63. [ Links ]
31. Mukherjee, A.; Vasquez, K. M. Triplex technology in studies of DNA damage, DNA repair, and mutagenesis. Biochimie 2011, 93, 1197-208. [ Links ]
32. Vasquez, K. M.; Glazer, P. M. Triplex-forming oligonucleotides: principles and applications. Quart. Rev. of Biophys. 2002, 35, 89-107. [ Links ]
33. Choi, J.; Majima, T. Conformational changes of non-B DNA. Chem. Soc. Rev. 2011, 40, 5893-909. [ Links ]
34. Berman, H. M.; Olson, W. K.; Beveridge, D. L.; Westbrook, J.; Gelbin, A.; Demeny, T.; Hsieh, S. H.; Srinivasan, A. R.; Schneider, B. The Nucleic Acid Database. A comprehensive relational database of three-dimensional structures of nucleic acids. Biophys. J. 1992, 63, 751-9. [ Links ]
35. Song, H.; Kaiser, J. T.; Barton, J. K. Crystal structure of Δ-[Ru(bpy)dppz]2+ bound to mismatched DNA reveals side-by-side metalloinsertion and intercalation. Nature Chemistry 2012, 4, 615-20. [ Links ]
36. Zhou, C.; Du, X.; Li, H. Studies of interactions among cobalt(III) polypyridyl complexes, 6-mercaptopurine and DNA. Bioelectrochem. 2007, 70, 446-51. [ Links ]
37. Wang, B.; Yang, Z.; Li, T. Synthesis, characterization, and DNA-binding properties of the Ln(III) complexes with 6-hydroxy chromone3-carbaldehyde-(2'-hydroxy) benzoyl hydrazone. Bioorg. Med. Chem. 2006, 14, 6012-21. [ Links ]
38. Calabrò, F.; Albers, P.; Bokemeyer, C.; Martin, C.; Einhorn, L. H.; Horwich, A.; Krege, S.; Schmoll, H. J.; Sternberg, C. N.; Daugaard, G. The contemporary role of chemotherapy for advanced testis cancer: a systematic review of the literature. Eur. Urol. 2012, 61, 1212-21. [ Links ]
39. Jamieson, E. R.; Lippard, S. J. Structure, Recognition, and Processing of Cisplatin–DNA Adducts. Chem. Rev. 1999, 99, 2467-2498. [ Links ]
40. Berners-Price, Sue; Appleton, T. G. The Chemistry of Cisplatin in Aqueous Solution. In Platinum-Based Drugs in Cancer Therapy; Humana Press: New Jersey, USA, 2000; pp. 3-35. [ Links ]
41. Kozelka, J.; Legendre, F.; Reeder, F.; Chottard, J.-C. Kinetic aspects of interactions between DNA and platinum complexes. Coord. Chem. Rev. 1999, 190-192, 61-82. [ Links ]
42. Davies, M. S.; Berners-Price, S. J.; Hambley, T. W. Slowing of Cisplatin Aquation in the Presence of DNA but Not in the Presence of Phosphate: Improved Understanding of Sequence Selectivity and the Roles of Monoaquated and Diaquated Species in the Binding of Cisplatin to DNA. Inorg. Chem. 2000, 39, 5603-5613. [ Links ]
43. Legendre, F.; Bas, V.; Kozelka, J.; Chottard, J. C. A complete kinetic study of GG versus AG plantination suggests that the doubly aquated derivatives of cisplatin are the actual DNA binding species. Chemistry-A Eur. J. 2000, 6, 2002-10. [ Links ]
44. Davies, M. S.; Berners-Price, S. J.; Hambley, T. W. Rates of Platination of AG and GA Containing Double-Stranded Oligonucleotides: Insights into Why Cisplatin Binds to GG and AG but Not GA Sequences in DNA. J. Am. Chem. Soc. 1998, 120, 11380-11390. [ Links ]
45. Berners-Price, S. J.; Frenkiel, T. A.; Frey, U.; Ranford, J. D.; Sadler, P. J. Hydrolysis products of cisplatin: pK a determinations via[1H, 15N] NMR spectroscopy. J. Chem. Soc. Chem. Commun. 1992, 789. [ Links ]
46. Eastman, A. The mechanism of action of cisplatin: from adducts to apoptosis. In Cisplatin: Chemistry and Biochemistry of a Leading Anticancer Drug; Lippert, B., Ed.; Verlag Helvetica Chimica Acta: Zurich, 1999; pp. 111-134. [ Links ]
47. Caradonna, J. P.; Lippard, S. J.; Gait, M. J.; Singh, M. The antitumor drug cis-dichlorodiammineplatinum forms an intrastrand d(GpG) crosslink upon reaction with [d(ApGpGpCpCpT)]2. J. Am. Chem. Soc. 1982, 104, 5793-5795. [ Links ]
48. Fichtinger-Schepman, A. M. J.; Van Oosterom, A. T.; Lohman, P. H. M.; Berends, F. cis-Diamminedichloroplatinum(II)-induced DNA Adducts in Peripheral Leukocytes from Seven Cancer Patients: Quantitative Immunochemical Detection of the Adduct Induction and Removal after a Single Dose of cis-Diamminedichloroplatinum(II). Cancer Res. 1987, 47, 3000-3004. [ Links ]
49. Fichtinger-Schepman, A. M. J.; Van der Veer, J. L.; Den Hartog, J. H. J.; Lohman, P. H. M.; Reedijk, J. Adducts of the antitumor drug cis-diamminedichloroplatinum(II) with DNA: formation, identification, and quantitation. Biochemistry 1985, 24, 707-713. [ Links ]
50. Filipski, J.; Kohn, K. W.; Bonner, W. M. The nature of inactivating lesions produced by platinum (II) complexes in phage λ DNA. Chemico-Biological Interactions 1980, 32, 321-330. [ Links ]
51. Eastman, A. Reevaluation of interaction of cis-dichloro(ethylenediamine)platinum(II) with DNA. Biochemistry 1986, 25, 3912-3915. [ Links ]
52. Hambley, T. W. Modelling the interaction of cisplatin with DNA. Drug Design and Delivery 1988, 3, 153-8. [ Links ]
53. Baik, M.-H.; Friesner, R. A.; Lippard, S. J. Theoretical study of cisplatin binding to purine bases: why does cisplatin prefer guanine over adenine? J. Am. Chem. Soc. 2003, 125, 14082-92. [ Links ]
54. Struik, A. Guanine-06 methylation reduces the reactivity of d(GpG) towards platinum complexes. J. Inorg. Biochem. 1991, 44, 249-260. [ Links ]
55. Hall, M. D.; Hambley, T. W. Platinum(IV) antitumour compounds: their bioinorganic chemistry. Coord. Chem. Rev. 2002, 232, 49-67. [ Links ]
56. Satraplatin: BMS 182751, BMY 45594, JM 216. Drugs in R & D 2007, 8, 125-32. [ Links ]
57. Lebwohl, D.; Canetta, R. Clinical development of platinum complexes in cancer therapy: an historical perspective and an update. Eur. J. Cancer 1998, 34, 1522-34. [ Links ]
58. Nordén, B.; Tjerneld, F. Binding of inert metal complexes to deoxyribonucleic acid detected by linear dichroism. FEBS Letters 1976, 67, 368-370. [ Links ]
59. Jennette, K. W.; Gill, J. T.; Sadownick, J. A.; Lippard, S. J. Metallointercalation reagents. Synthesis, characterization, and structural properties of thiolato(2,2',2''-terpyridine)platinum(II) complexes. J. Am. Chem. Soc. 1976, 98, 6159-6168. [ Links ]
60. Yang, D.; Van Boom, S. S.; Reedijk, J.; Van Boom, J. H.; Farrell, N.; Wang, A. H. A novel DNA structure induced by the anticancer bisplatinum compound crosslinked to a GpC site in DNA. Nature Struct. Biol. 1995, 2, 577-8. [ Links ]
61. Farrell, N.; Appleton, T. G.; Qu, Y.; Roberts, J. D.; Fontes, A. P. S.; Skov, K. A.; Wu, P.; Zou, Y. Effects of Geometric Isomerism and Ligand Substitution in Bifunctional Dinuclear Platinum Complexes on Binding Properties and Conformational Changes in DNA. Biochemistry 1995, 34, 15480-15486. [ Links ]
62. Roberts, J. D.; Van Houten, B.; Qu, Y.; Farrell, N. P. Interaction of novel bis(platinum) complexes with DNA. Nuc. Ac. Res. 1989, 17, 9719-9733. [ Links ]
63. Kasparkova, J.; Zehnulova, J.; Farrell, N.; Brabec, V. DNA interstrand cross-links of the novel antitumor trinuclear platinum complex BBR3464. Conformation, recognition by high mobility group domain proteins, and nucleotide excision repair. J. Biol. Chem. 2002, 277, 48076-86. [ Links ]
64. Noll, D. M.; Mason, T. M.; Miller, P. S. Formation and repair of interstrand cross-links in DNA. Chem. Rev. 2006, 106, 277-301. [ Links ]
65. Rajski, S. R.; Williams, R. M. DNA Cross-Linking Agents as Antitumor Drugs. Chem. Rev. 1998, 98, 2723-2796. [ Links ]
66. Roberts, J. D.; Peroutka, J.; Farrell, N. Cellular pharmacology of polynuclear platinum anti-cancer agents. J. Inorg. Biochem. 1999, 77, 51-7. [ Links ]
67. Wheate, N. Multi-nuclear platinum complexes as anti-cancer drugs. Coord. Chem. Rev. 2003, 241, 133-145. [ Links ]
68. Gourley, C.; Cassidy, J.; Edwards, C.; Samuel, L.; Bisset, D.; Camboni, G.; Young, A.; Boyle, D.; Jodrell, D. A phase I study of the trinuclear platinum compound, BBR 3464, in combination with protracted venous infusional 5-fluorouracil in patients with advanced cancer. Cancer Chemother. Pharmacol. 2004, 53, 95-101. [ Links ]
69. Boulikas, T.; Vougiouka, M. Cisplatin and platinum drugs at the molecular level. (Review). Oncology Rep. 2003, 10, 1663-82. [ Links ]
70. Roberts, J. D.; Peroutka, J.; Beggiolin, G.; Manzotti, C.; Piazzoni, L.; Farrell, N. Comparison of cytotoxicity and cellular accumulation of polynuclear platinum complexes in L1210 murine leukemia cell lines. J. Inorg. Biochem. 1999, 77, 47-50. [ Links ]
71. Perego, P.; Gatti, L.; Caserini, C.; Supino, R.; Colangelo, D.; Leone, R.; Spinelli, S.; Farrell, N.; Zunino, F. The cellular basis of the efficacy of the trinuclear platinum complex BBR 3464 against cisplatin-resistant cells. J. Inorg. Biochem. 1999, 77, 59-64. [ Links ]
72. Perego, P.; Caserini, C.; Gatti, L.; Carenini, N.; Romanelli, S.; Supino, R.; Colangelo, D.; Viano, I.; Leone, R.; Spinelli, S.; Pezzoni, G.; Manzotti, C.; Farrell, N.; Zunino, F. A novel trinuclear platinum complex overcomes cisplatin resistance in an osteosarcoma cell system. Molec. Pharmacol. 1999, 55, 528-34. [ Links ]
73. Manzotti, C.; Pratesi, G.; Menta, E.; Di Domenico, R.; Cavalletti, E.; Fiebig, H. H.; Kelland, L. R.; Farrell, N.; Polizzi, D.; Supino, R.; Pezzoni, G.; Zunino, F. BBR 3464: a novel triplatinum complex, exhibiting a preclinical profile of antitumor efficacy different from cisplatin. Clin. Cancer Res. 2000, 6, 2626-34. [ Links ]
74. Hensing, T. A.; Hanna, N. H.; Gillenwater, H. H.; Gabriella Camboni, M.; Allievi, C.; Socinski, M. A. Phase II study of BBR 3464 as treatment in patients with sensitive or refractory small cell lung cancer. Anti-Cancer Drugs 2006, 17, 697-704. [ Links ]
75. Jodrell, D. I.; Evans, T. R. J.; Steward, W.; Cameron, D.; Prendiville, J.; Aschele, C.; Noberasco, C.; Lind, M.; Carmichael, J.; Dobbs, N.; Camboni, G.; Gatti, B.; De Braud, F. Phase II studies of BBR3464, a novel tri-nuclear platinum complex, in patients with gastric or gastro-oesophageal adenocarcinoma. Eur. J. Cancer 2004, 40, 1872-7. [ Links ]
76. Gessner, R. V; Quigley, G. J.; Wang, A. H.; Van der Marel, G. A.; Van Boom, J. H.; Rich, A. Structural basis for stabilization of Z-DNA by cobalt hexaammine and magnesium cations. Biochemistry 1985, 24, 237-40. [ Links ]
77. Mei, H. Y.; Barton, J. K. Tris(tetramethylphenanthroline)ruthenium(II): a chiral probe that cleaves A-DNA conformations. Proc. Nat. Ac. Sci. 1988, 85, 1339-43. [ Links ]
78. Mariappan, M.; Suenaga, M.; Mukhopadhyay, A.; Raghavaiah, P.; Maiya, B. G. Synthesis, structure, DNA binding and photocleavage activity of a ruthenium(II) complex with 11-(9-acridinyl)dipyrido[3,2-a:2',3'-c]phenazine ligand. Inorg. Chim. Acta 2011, 376, 340-349. [ Links ]
79. Maity, B.; Roy, M.; Chakravarty, A. R. Ferrocene-conjugated copper(II) dipyridophenazine complex as a multifunctional model nuclease showing DNA cleavage in red light. J. Organomet. Chem. 2008, 693, 1395-1399. [ Links ]
80. Hannon, M. J.; Moreno, V.; Prieto, M. J.; Moldrheim, E.; Sletten, E.; Meistermann, I.; Isaac, C. J.; Sanders, K. J.; Rodger, A. Intramolecular DNA Coiling Mediated by a Metallo-Supramolecular Cylinder. Angew. Chem. Int. Ed. 2001, 40, 879-884. [ Links ]
81. Meistermann, I.; Moreno, V.; Prieto, M. J.; Moldrheim, E.; Sletten, E.; Khalid, S.; Rodger, P. M.; Peberdy, J. C.; Isaac, C. J.; Rodger, A.; Hannon, M. J. Intramolecular DNA coiling mediated by metallo-supramolecular cylinders: differential binding of P and M helical enantiomers. Proc. Nat. Ac. Sci. USA 2002, 99, 5069-74. [ Links ]
82. Peberdy, J. C.; Malina, J.; Khalid, S.; Hannon, M. J.; Rodger, A. Influence of surface shape on DNA binding of bimetallo helicates. J. Inorg. Biochem. 2007, 101, 1937-45. [ Links ]
83. McDonnell, U.; Hicks, M. R.; Hannon, M. J.; Rodger, A. DNA binding and bending by dinuclear complexes comprising ruthenium polypyridyl centres linked by a bis(pyridylimine) ligand. J. Inorg. Biochem. 2008, 102, 2052-9. [ Links ]
84. Lerman, L. S. Structural considerations in the interaction of DNA and acridines. J. Molec. Biol. 1961, 3, 18-30. [ Links ]
85. Dupureur, C. M.; Barton, J. K. Use of Selective Deuteration and 1H NMR in Demonstrating Major Groove Binding of .DELTA.[Ru(phen)2dppz]2+ to d(GTCGAC)2. J. Am. Chem. Soc. 1994, 116, 10286-10287. [ Links ]
86. Greguric, A.; Greguric, I. D.; Hambley, T. W.; Aldrich-Wright, J. R.; Collins, J. G. Minor groove intercalation of Δ-[Ru(Me2phen)2dppz]2+ to the hexanucleotide d(GTCGAC)2. Dalton Transactions 2002, 849. [ Links ]
87. Pindur, U.; Haber, M.; Sattler, K. Antitumor active drugs as intercalators of deoxyribonucleic acid: Molecular models of intercalation complexes. J. Chem. Ed. 1993, 70, 263. [ Links ]
88. Berman, H. M.; Young, P. R. The interaction of intercalating drugs with nucleic acids. Ann. Rev. Biophys. Bioeng. 1981, 10, 87-114. [ Links ]
89. Howe-Grant, M.; Wu, K. C.; Bauer, W. R.; Lippard, S. J. Binding of platinum and palladium metallointercalation reagents and antitumor drugs to closed and open DNAs. Biochemistry 1976, 15, 4339-4346. [ Links ]
90. Lippard, S.; Bond, P.; WU, K.; Bauer, W. Stereochemical requirements for intercalation of platinum complexes into double-stranded DNA's. Science 1976, 194, 726-728. [ Links ]
91. Barton, J. K.; Dannenberg, J. J.; Raphael, A. L. Enantiomeric selectivity in binding tris(phenanthroline)zinc(II) to DNA. J. Am. Chem. Soc. 1982, 104, 4967-4969. [ Links ]
92. Stinner, C.; Wightman, M. D.; Kelley, S. O.; Hill, M. G.; Barton, J. K. Synthesis and Spectroelectrochemistry of Ir(bpy)(phen)(phi) 3+ , a Tris(heteroleptic) Metallointercalator. Inorg. Chem. 2001, 40, 5245-5250. [ Links ]
93. Carter, M. T.; Rodriguez, M.; Bard, A. J. Voltammetric studies of the interaction of metal chelates with DNA. 2. Tris-chelated complexes of cobalt(III) and iron(II) with 1,10-phenanthroline and 2,2'-bipyridine. J. Am. Chem. Soc. 1989, 111, 8901-8911. [ Links ]
94. Yoshioka, N.; Inoue, H. DNA binding of iron(II) mixed-ligand complexes containing 1,10-phenanthroline and 4,7-diphenyl-1,10-phenanthroline. J. Inorg. Biochem. 1999, 77, 239-247. [ Links ]
95. Arounaguiri, S.; Maiya, B. G. Dipyridophenazine Complexes of Cobalt(III) and Nickel(II): DNA-Binding and Photocleavage Studies. Inorg. Chem. 1996, 35, 4267-4270. [ Links ]
96. Barton, J. K.; Raphael, A. L. Photoactivated stereospecific cleavage of double-helical DNA by cobalt(III) complexes. J. Am. Chem. Soc. 1984, 106, 2466-2468. [ Links ]
97. Holmlin, R. E.; Yao, J. A.; Barton, J. K. Dipyridophenazine Complexes of Os(II) as Red-Emitting DNA Probes: Synthesis, Characterization, and Photophysical Properties. Inorg. Chem. 1999, 38, 174-189. [ Links ]
98. Holmlin, R. E.; Barton, J. K. Os(phen)2(dppz)2+: A Red-Emitting DNA Probe. Inorg. Chem. 1995, 34, 7-8. [ Links ]
99. Zeglis, B. M.; Barton, J. K. DNA base mismatch detection with bulky rhodium intercalators: synthesis and applications. Nature Protocols 2007, 2, 357-71. [ Links ]
100. Arkin, M. R.; Stemp, E. D. A.; Holmlin, R. E.; Barton, J. K.; Hormann, A.; Olson, E. J. C.; Barbara, P. F. Rates of DNA-Mediated Electron Transfer Between Metallointercalators. Science 1996, 273, 475-480. [ Links ]
101. Erkkila, K. E.; Odom, D. T.; Barton, J. K. Recognition and Reaction of Metallointercalators with DNA. Chem. Rev. 1999, 99, 2777-2796. [ Links ]
102. Neidle, S.; Abraham, Z. Structural and sequence-dependent aspects of drug intercalation into nucleic acids. Crit. Rev. Biochem. 1984, 17, 73-121. [ Links ]
103. Sigman, D. S. Chemical nucleases. Biochemistry 1990, 29, 9097-9105. [ Links ]
104. Brodie, C. R.; Collins, J. G.; Aldrich-Wright, J. R. DNA binding and biological activity of some platinum(II) intercalating compounds containing methyl-substituted 1,10-phenanthrolines. Dalton Transactions 2004, 1145-52. [ Links ]
105. McFadyen, W. D.; Wakelin, L. P. G.; Roos, I. A. G.; Leopold, V. A. Activity of platinum(II) intercalating agents against murine leukemia L1210. J. Med. Chem. 1985, 28, 1113-1116. [ Links ]
106. Che, C.-M.; Yang, M.; Wong, K.-H.; Chan, H.-L.; Lam, W. Platinum(II) Complexes of Dipyridophenazine as Metallointercalators for DNA and Potent Cytotoxic Agents against Carcinoma Cell Lines. Chemistry, Eur. J. 1999, 5, 3350-3356. [ Links ]
107. McCoubrey, A.; Latham, H. C.; Cook, P. R.; Rodger, A.; Lowe, G. 4-Picoline-2,2':6',2"-terpyridine-platinum(II) - A potent intercalator of DNA. FEBS Letters 1996, 380, 73-78. [ Links ]
108. Corral, E.; Hotze, A. C. G.; Den Dulk, H.; Leczkowska, A.; Rodger, A.; Hannon, M. J.; Reedijk, J. Ruthenium polypyridyl complexes and their modes of interaction with DNA: is there a correlation between these interactions and the antitumor activity of the compounds? J. Biol. Inorg. Chem. 2009, 14, 439-48. [ Links ]
109. Chikira, M.; Tomizawa, Y.; Fukita, D.; Sugizaki, T.; Sugawara, N.; Yamazaki, T.; Sasano, A.; Shindo, H.; Palaniandavar, M.; Antholine, W. DNA-fiber EPR study of the orientation of Cu(II) complexes of 1,10-phenanthroline and its derivatives bound to DNA: mono(phenanthroline)-copper(II) and its ternary complexes with amino acids. J. Inorg. Biochem. 2002, 89, 163-173. [ Links ]
110. Bravo-Gómez, M. E.; García-Ramos, J. C.; Gracia-Mora, I.; Ruíz-Azuara, L. Antiproliferative activity and QSAR study of copper(II) mixed chelate [Cu(N-N)(acetylacetonato)]NO3 and [Cu(N-N)(glycinato)]NO3 complexes, (Casiopeínas). J. Inorg. Biochem. 2009, 103, 299-309. [ Links ]
111. Vagg, R. S.; Williams, P. A. Chiral metal complexes 2. Light-catalysed diastereoisomeric equilibration in aqueous solutions of cis[Ru(phen)2(L-serine)]+ and its 2,2'-bipyridyl analogue. Inorg. Chim. Acta 1981, 52, 69-72. [ Links ]
112. Sardesai, N. Y.; Lin, S. C.; Zimmermann, K.; Barton, J. K. Construction of Coordinatively Saturated Rhodium Complexes Containing Appended Peptides. Bioconj. Chem. 1995, 6, 302-312. [ Links ]
113. Rutherford, T. J.; Quagliotto, M. G.; Keene, F. R. Chiral [Ru(pp)2(CO)2]2+ Species (pp = Bidentate Polypyridyl Ligand) and Their Use in the Stereoselective Synthesis of Ligand-Bridge Dinuclear Complexes. Inorg. Chem. 1995, 34, 3857-3858. [ Links ]
114. Hua, X.; Von Zelewsky, A. Enantiomerically Pure Chiral RuII(L-L)2 Building Blocks for Coordination Compounds. Inorg. Chem. 1995, 34, 5791-5797. [ Links ]
115. Helms, J. B.; Huang, L.; Price, R.; Sullivan, B. P.; Sullivan, B. A. Coordination Chemistry of Tropolone-Based Antimitotic Drugs and the Antineoplastic Behavior of Some Ruthenium(II) and Platinum(II) Derivatives. Inorg. Chem. 1995, 34, 5335-5340. [ Links ]
116. Sitlani, A.; Barton, J. K. Sequence-specific recognition of DNA by phenanthrenequinone diimine complexes of rhodium(III): importance of steric and van der Waals interactions. Biochemistry 1994, 33, 12100-8. [ Links ]
117. Sardesai, N. Y.; Zimmermann, K.; Barton, J. K. DNA Recognition by Peptide Complexes of Rhodium(III): Example of a Glutamate Switch. J. Am. Chem. Soc. 1994, 116, 7502-7508. [ Links ]
118. Sitlani, A.; Dupureur, C. M.; Barton, J. K. Enantiospecific palindromic recognition of 5'-d(CTCTAGAG)-3' by a novel rhodium intercalator: analogies to a DNA-binding protein. J. Am. Chem. Soc. 1993, 115, 12589-12590. [ Links ]
119. Satyanarayana, S.; Dabrowiak, J. C.; Chaires, J. B. Tris(phenanthroline)ruthenium(II) enantiomer interactions with DNA: mode and specificity of binding. Biochemistry 1993, 32, 2573-84. [ Links ]
120. Murphy, C. J.; Barton, J. K. Ruthenium complexes as luminescent reporters of DNA. Meth. Enzymol. 1993, 226, 576-94. [ Links ]
121. Togano, T.; Nagao, N.; Tsuchida, M.; Kumakura, H.; Hisamatsu, K.; Howell, F. S.; Mukaida, M. One-pot and selective synthesis of a series of [RuCl6−2nLn] (L=bidentate ligand, n=0−3) types of complexes with polypyridyl ligands; another example of the synthetic utility of "ruthenium-blue" solution. Inorg. Chim. Acta 1992, 195, 221-225. [ Links ]
122. Sitlani, A.; Long, E. C.; Pyle, A. M.; Barton, J. K. DNA photocleavage by phenanthrenequinone diimine complexes of rhodium(III): shape-selective recognition and reaction. J. Am. Chem. Soc. 1992, 114, 2303-2312. [ Links ]
123. Satyanarayana, S.; Dabrowiak, J. C.; Chaires, J. B. Neither delta-nor lambda-tris(phenanthroline)ruthenium(II) binds to DNA by classical intercalation. Biochemistry 1992, 31, 9319-24. [ Links ]
124. Hartshorn, R. M.; Barton, J. K. Novel dipyridophenazine complexes of ruthenium(II): exploring luminescent reporters of DNA. J. Am. Chem. Soc. 1992, 114, 5919-5925. [ Links ]
125. Peek, B. M.; Ross, G. T.; Edwards, S. W.; Meyer, G. J.; Meyer, T. J.; Erickson, B. W. Synthesis of redox derivatives of lysine and related peptides containing phenothiazine or tris(2,2'-bipyridine)ruthenium(II). Int. J. Peptide Prot. Res. 1991, 38, 114-23. [ Links ]
126. Morgan, R. J.; Chatterjee, S.; Baker, A. D.; Strekas, T. C. Effects of ligand planarity and peripheral charge on intercalative binding of Ru(2,2'-bipyridine)2L2+ to calf thymus DNA. Inorg. Chem. 1991, 30, 2687-2692. [ Links ]
127. Friedman, A. E.; Kumar, C. V; Turro, N. J.; Barton, J. K. Luminescence of ruthenium(II) polypyridyls: evidence for intercalative binding to Z-DNA. Nuc. Ac. Res. 1991, 19, 2595-602. [ Links ]
128. Rehmann, J. P.; Barton, J. K. 1H NMR studies of tris(phenanthroline) metal complexes bound to oligonucleotides: characterization of binding modes. Biochemistry 1990, 29, 1701-9. [ Links ]
129. Patra, A. K.; Rose, M. J.; Murphy, K. A.; Olmstead, M. M.; Mascharak, P. K. Photolabile ruthenium nitrosyls with planar dicarboxamide tetradentate N(4) ligands: effects of in-plane and axial ligand strength on NO release. Inorg. Chem. 2004, 43, 4487-95. [ Links ]
130. Friedman, A. E.; Chambron, J. C.; Sauvage, J. P.; Turro, N. J.; Barton, J. K. A molecular light switch for DNA: Ru(bpy)2(dppz)2+. J. Am. Chem. Soc. 1990, 112, 4960-4962. [ Links ]
131. Cathey, C. J.; Constable, E. C.; Hannon, M. J.; Tohcer, D. A.; Ward, M. D. A single stranded diruthenium(II) helical complex. J. Chem. Soc. Chem. Commun. 1990, 621. [ Links ]
132. De Buyl, F.; Kirsch-De Mesmaeker, A.; Tossi, A.; Kelly, J. M. Medium dependence of the spectroscopic and photophysical properties of Ru(bpy)2(HAT)2+. The effect of solvent, pH and binding to polyelectrolytes. J. Photochem. Photobiol. A: Chemistry 1991, 60, 27-45. [ Links ]
133. Baker, A. D.; Morgan, R. J.; Strekas, T. C. Enantiospecific cleavage of DNA using copper(II) chelated to the periphery of a ligand on a chiral tris-chelate of ruthenium(II). J. Chem. Soc. Chem. Commun. 1992, 1099. [ Links ]
134. Baker, A. D.; Morgan, R. J.; Strekas, T. C. Enantiomeric resolution of ruthenium complexes Ru(phen)32+ and Ru(bpy)2ppz2+ on a DNA-hydroxylapatite column. J. Am. Chem. Soc. 1991, 113, 1411-1412. [ Links ]
135. Vagg, R. S.; Williams, P. A. Chiral metal complexes. 1. Photochemical inversion in ternary Ru(II) complexes of diimines and L-tryptophane. Inorg. Chim. Acta 1981, 51, 61-65. [ Links ]
136. Nordén, B.; Kurucsev, T. Analysing DNA complexes by circular and linear dichroism. J. Mol. Recogn. 1994, 7, 141-55. [ Links ]
137. Eriksson, M.; Leijon, M.; Hiort, C.; Nordén, B.; Gräslund, A. Binding of delta- and lambda-[Ru(phen)3]2+ to [d(CGCGATCGCG)]2 studied by NMR. Biochemistry 1994, 33, 5031-40. [ Links ]
138. Hiort, C.; Lincoln, P.; Norden, B. DNA binding of .DELTA.- and .LAMBDA.-[Ru(phen)2DPPZ]2+. J. Am. Chem. Soc. 1993, 115, 34483454. [ Links ]
139. Eriksson, M.; Leijon, M.; Hiort, C.; Norden, B.; Gräslund, A. Minor groove binding of [Ru(phen)3]2+ to [d(CGCGATCGCG)]2 evidenced by two-dimensional NMR. J. Am. Chem. Soc. 1992, 114, 4933-4934. [ Links ]
140. Hiort, C.; Norden, B.; Rodger, A. Enantiopreferential DNA binding of [ruthenium(II)(1,10-phenanthroline)3]2+ studied with linear and circular dichroism. J. Am. Chem. Soc. 1990, 112, 1971-1982. [ Links ]
141. Dwyer, F. p.; gyarfas, e. c.; rogers, w. p.; koch, j. h. Biological Activity of Complex Ions. Nature 1952, 170, 190-191. [ Links ]
142. Barton, J. K. Tris (phenanthroline) metal complexes: probes for DNA helicity. J. Biomol. Struct. Dynam. 1983, 1, 621-32. [ Links ]
143. Barton, J. K.; Basile, L. A.; Danishefsky, A.; Alexandrescu, A. Chiral probes for the handedness of DNA helices: enantiomers of tris(4,7diphenylphenanthroline)ruthenium(II). Proc. Nat. Acad. Sci. USA 1984, 81, 1961-5. [ Links ]
144. Barton, J. K.; Goldberg, J. M.; Kumar, C. V.; Turro, N. J. Binding modes and base specificity of tris(phenanthroline)ruthenium(II) enantiomers with nucleic acids: tuning the stereoselectivity. J. Am. Chem. Soc. 1986, 108, 2081-2088. [ Links ]
145. Coggan, D. Z. M.; Haworth, I. S.; Bates, P. J.; Robinson, A.; Rodger, A. DNA Binding of Ruthenium Tris(1,10-phenanthroline): Evidence for the Dependence of Binding Mode on Metal Complex Concentration. Inorg. Chem. 1999, 38, 4486-4497. [ Links ]
146. Wheate, N. J.; Brodie, C. R.; Collins, J. G.; Kemp, S.; Aldrich-Wright, J. R. DNA intercalators in cancer therapy: organic and inorganic drugs and their spectroscopic tools of analysis. Mini Rev. Med. Chem. 2007, 7, 627-48. [ Links ]
147. Lowe, G.; Droz, A. S.; Vilaivan, T.; Weaver, G. W.; Park, J. J.; Pratt, J. M.; Tweedale, L.; Kelland, L. R. Cytotoxicity of 2,2':6',2''terpyridineplatinum(II) complexes against human ovarian carcinoma. J. Med. Chem. 1999, 42, 3167-74. [ Links ]
148. Sitlani, A.; Dupureur, C. M.; Barton, J. K. Enantiospecific palindromic recognition of 5'-d(CTCTAGAG)-3' by a novel rhodium intercalator: analogies to a DNA-binding protein. J. Am. Chem. Soc. 1993, 115, 12589-12590. [ Links ]
149. Sitlani, A.; Barton, J. K. Sequence-Specific Recognition of DNA by Phenanthrenequinone Diimine Complexes of Rhodium(III): Importance of Steric and van der Waals Interactions. Biochemistry 1994, 33, 12100-12108. [ Links ]
150. Kang, M.; Chouai, A.; Chifotides, H. T.; Dunbar, K. R. 2D NMR Spectroscopic Evidence for Unprecedented Interactions of Cis- Rh2(dap)( μ -O2CCH3)2(μ 1-O2CCH3)(CH3OH) (O2CCH3) with a DNA Oligonucleotide: Combination of Intercalative and Coordinative Binding. Angew. Chem. Int. Ed. 2006, 45, 6148-6151. [ Links ]
151. Frodl, A.; Herebian, D.; Sheldrick, W. S. Coligand Tuning of the DNA Binding Properties of Bioorganometallic (η6-arene) Ruthenium(II) Complexes of the Type [(η6-arene)Ru(amino acid)(dppz)]n+ (dppz = dipyrido[3,2-a:2',3'-c]phenazine), n=1-3. J. Chem. Soc. Dalton Trans. 2002, 3664-3673. [ Links ]
152. Escribano, E.; Font-Bardia, M.; Calvet, T.; Lorenzo, J.; Gamez, P.; Moreno. V. DNA binding studies of a series of cis-[Pt(Am)2X2] complexes (Am = inert amine, X = labile carboxylato ligand) Inorg. Chim. Acta 2013, 394, 65-76. [ Links ]
153. Ruiz, J.; Cutillas, N.; Vicente, C.; Villa, M.D.; López, G.; Lorenzo, J.; Avilés, F.X.; Moreno, V.; Bautista, D. New Palladium(II) and Platinum(II) Complexes with the Model Nucleobase 1-Methylcytosine: Antitumor Activity and Interactions with DNA. Inorg. Chem. 2005, 44 (21), 365-7376. [ Links ]
154. Albert, J.; García, S.; Granell, J.; Llorca, A.; Lovelle, M.V.; Moreno, V.; Presa, A.; Rodríguez, L.; Quirante, J.; Calvis, C.; Messeguer, R.; Badía, J.; Baldomà, L. Cyclopalladated benzophenone imines: Synthesis, cytotoxicity against human breast adenocarcinoma cell lines and DNA interaction. J. Organomet. Chem. 2013, 724, 289-296. [ Links ]
155. Garcia, M.H.; Valente, A.; Florindo, P.; Morais, T.S.; Piedade,M.F.M.; Duarte, M.T.; Moreno, V.; Avilés, F.X.; Loreno, J. New ruthenium(II) mixed metallocene derived complexes: Synthesis, characterization by X-ray diffraction and evaluation on DNA interaction by atomic force microscopy Inorg. Chim. Acta 2013, 363(14), 3765-3775. [ Links ]
156. Moreno, V.; Font-Bardia, M. Calvet, T.; Lorenzo, J.; Avilés, F.X.; Garcia, M.H.; Morais, T.S.; Valente, A.; Robalo, M.P. DNA interaction and cytotoxicity studies of new ruthenium(II) cyclopentadienyl derivative complexes containing heteroaromatic ligands. J. Inorg. Biochem. 2011, 105(2), 241-249. [ Links ]
157. Barragán,F.; Carrion-Salip, D.; Gómez-Pinto, I.; González-Cantó, A.; Sadler, P.J.; de Llorens,R.; Moreno,V.; González, C.; Massaguer, A.; Marchán, V. Somatostatin Subtype-2 Receptor-Targeted Metal-Based Anticancer Complexes Bioconjugate Chem., 2012, 23 (9), 1838-1855. [ Links ]
158. Gómez-Segura,J.; Prieto, M.J.; Font-Bardia,M.; Solans,X.; Moreno, V. Crystal Structure and DNA Interaction of the Facial-Type rac-Tris[2-(aminomethyl)benzimidazole]cobalt(III) Complex. Inorg. Chem. 2006, 45 (25), 10031-10033. [ Links ]
159. Arbuse, A.; Font, M.; Martínez, M.A., Fontrodona, X.; Prieto, M.J.; Moreno, V.; Sala, X.; Llobet, A. DNA-Cleavage Induced by New Macrocyclic Schiff base Dinuclear Cu(I) Complexes Containing Pyridyl Pendant Arms. Inorg. Chem. 2009, 48 (23), 11098-11107. [ Links ]
160. Becco, L.; Rodríguez, A.; Bravo, M.E.; Prieto, M.J.; Ruiz-Azuara, L.; Garat, B.; Moreno, V.; Gambino, D. New achievements on biological aspects of copper complexes Casiopeínas®: Interaction with DNA and proteins and anti-Trypanosoma cruzi activity. J. Inorg. Biochem. 2012, 109, 49-56. [ Links ]
161. Benítez, J.; Cavalcanti de Queiroz, A.; Correia,I.; Amaral Alves, M.; Alexandre-Moreira, M.S.; Barreiro, E.J.; Moreira Lima, L.; Varela, J.; González, M.; Cerecetto, H.; Moreno, V.; Costa Pessoa, J.; Gambino, D. New oxidovanadium(IV) N-acylhydrazone complexes: Promising antileishmanial and antitrypanosomal agents. Eur. J. Med. Chem. 2013, 62, 20-27. [ Links ]
162. Fernández, M.; Becco, L.; Correia, I.; Benítez, J.; Piro, O.E., Echeverria, G.A.; Medeiros, A.; Comini, M.; Lavaggi, M.L., González, M.; Cerecetto, H., Moreno, V.; Costa Pessoa, J.; Garat, B.; Gambino, D. Oxidovanadium(IV) and dioxidovanadium(V) complexes of tridentate salicylaldehyde semicarbazones: Searching for prospective antitrypanosomal agents J. Inorg. Biochem. doi: http://dx.doi.org/10.1016/j.jinorgbio.2013.02.010 [ Links ]
163. Benítez, J.; Becco, L.; Correia, I.; Leal, S.M.; Guiset, H.; Costa Pessoa, J.; Lorenzo, J.; Tanco, S.; Escobar, P.; Moreno, V., Garat, B.; Gambino, D. Vanadium polypyridyl compounds as potential antiparasitic and antitumoral agents: New achievements J. Inorg. Biochem., 2011, 105, 303-312. [ Links ]
164. Vieites, M.; Smircich, P.; Pagano, M.; Otero, L.; Luane Fischer, F.; Terenzi, H., Prieto, M.J., Moreno, V.; Garat, B., Gambino, D. DNA as molecular target of analogous palladium and platinum anti-Trypanosoma cruzi compounds: A comparative study. J. Inorg. Biochem. 2011, 105, 1704-1711. [ Links ]
165. Pavan, F.R.; Poelhsitz, G.V.; Barbosa, M.I.F.; Leite, S.R.A.; Batista, A.A.; Ellena, J.; Sato, L.S.; Franzblau, S.G.; Moreno, V.; Gambino, D.; Leite, C.Q.F. Ruthenium(II) phosphine/diimine/picolinate complexes: Inorganic compounds as agents against tuberculosis. Eur. J. Med. Chem. 2011, 46, 5099-5107. [ Links ]
166. Navarro, M.; Hernández, C.; Colmenares, I.; Hernández, P., Fernández, M.; Sierraalta, A.; Marchán, E. Synthesis and characterization of [Au(dppz)2]Cl3. DNA interaction studies and biological activity against Leishmania (L) mexicana J. Inorg. Biochem. 2007, 101, 111-116. [ Links ]
167. Navarro, M.; Cisneros-Fajardo, E.J.; Fernandez-Mestre, M.; Arrieche, D.; Marchán, E. Synthesis, characterization, DNA binding study and biological activity against Leishmania mexicana of [Cu(dppz)2]BF4 J. Inorg. Biochem. 2003, 97, 364-369 [ Links ]
168. Navarro, M.; Castro, W.; Higuera-Padilla, A.R.; Sierraalta, A.; Abad, M.J.; Taylor, P.; Sánchez-Delgado, R.A. Synthesis, characterization and biological activity of trans-platinum(II) complexes with chloroquine. J. Inorg. Biochem., 2011, 105, 1684-1691. [ Links ]
169. Sponer, J.; Burda, J. V.; Leszczynski, J. Interactionf of Metal Cations with Nucleic Acids and their building units. In Computational Studies of RNA and DNA; Šponer, J.; Lankaš, F., Eds.; Springer Netherlands, 2006; pp. 389-409. [ Links ]
170. Pérez, A.; Luque, F. J.; Orozco, M. Dynamics of B-DNA on the microsecond time scale. J. Am. Chem. Soc. 2007, 129, 14739-45. [ Links ]
171. Henriksen, N. M.; Davis, D. R.; T. E. Cheatham, I. Molecular dynamics re-refinement of two different small RNA loop structures using the original NMR data suggest a common structure. J. Biomol. NMR 2012, 53, 321-39. [ Links ]
172. Young, M. A.; Ravishanker, G.; Beveridge, D. L. A 5-nanosecond molecular dynamics trajectory for B-DNA: analysis of structure, motions, and solvation. Biophys. J. 1997, 73, 2313-36. [ Links ]
173. Yue, H.; Zhu, Y.; Wang, Y.; Chen, G. Investigation and improvement of DNA cleavage models of polyamide + Cu(II) nuclease + OOH- ligands bound to DNA. BMC structural biology 2010, 10, 35. [ Links ]
174. Sekharudu, C. Y.; Yathindra, N.; Sundaralingam, M. Molecular dynamics investigations of DNA triple helical models: unique features of the Watson-Crick duplex. J. Biomol. Struct. Dynam. 1993, 11, 225-44. [ Links ]
175. Wu, L.; Reymer, A.; Persson, C.; Kazimierczuk, K.; Brown, T.; Lincoln, P.; Nordén, B.; Billeter, M. Initial DNA Interactions of the Binuclear Threading Intercalator Λ,Λ-[μ-bidppz(bipy)4 Ru2](4+): An NMR Study with [d(CGCGAATTCGCG)]2. Chemistry, Eur. J. 2013, 19, 5401-5410. [ Links ]
176. Peters, M. B.; Yang, Y.; Wang, B.; Füsti-Molnár, L.; Weaver, M. N.; Merz, K. M. Structural Survey of Zinc Containing Proteins and the Development of the Zinc AMBER Force Field (ZAFF). J. Chem. Theo. Comput. 2010, 6, 2935-2947. [ Links ]
177. Hoops, S. C.; Anderson, K. W.; Merz, K. M. Force field design for metalloproteins. J. Am. Chem. Soc. 1991, 113, 8262-8270. [ Links ]
178. Case, D. A.; Cheatham, T. E. I.; Darden, T.; Gohlke, H.; Luo, R.; Merz, K. M.; Onufriev, A.; Simmerling, C.; Wang, B.; Woods, R. J. The Amber biomolecular simulation programs. J. Computat. Chem. 2005, 26, 1668-88. [ Links ]
179. Babu, C. S.; Lim, C. Empirical force fields for biologically active divalent metal cations in water. J. Phys. Chem. A 2006, 110, 691-9. [ Links ]
180. Ruiz-Azuara, L. L. Ruiz-Azuara,. SECOFI Patente, 26/01/1994. 1996; 21/04/1992 U.S. Patent No. 5, 107, 005. Re35, 458, Feb. 18 (1997); L. Ruiz-Azuara, SECOFI (1990).Patente, 09/12/1993 no. 172248. 1997: U.S. Patent No.5,576,326. 19/11/1996. [ Links ]
181. R. Galindo-Murillo, L. Ruiz-Azuara, R. Moreno-Esparza, and F. Cortés-Guzmán, Molecular recognition between DNA and a copper-based anticancer complex. Phys. Chem. Chem. Phys. 2012, 14, 15539-46. [ Links ]
182. Bayly, C. I.; Cieplak, P.; Cornell, W. D.; Kollman, P. A. A well-behaved electrostatic potential based method using charge restraints for deriving atomic charges: the RESP model. J. Phys. Chem. 1993, 97, 10269-10280. [ Links ]
183. Matsubara, T.; Hirao, K. Density functional study of the binding of the cyclen-coordinated M(II) (M=Zn, Cu, Ni) complexes to the DNA base. Why is Zn better to bind? J. Molec. Struct. THEOCHEM 2002, 581, 203-213. [ Links ]
184. Li, J.; Chen, J.; Xu, L.; Zheng, K.; Ji, L. A DFT/TDDFT study on the structures, trend in DNA-binding and spectral properties of molecular "light switch" complexes [Ru(phen)2(L)]2+(L=dppz, taptp, phehat). J. Organomet. Chem. 2007, 692, 831-838. [ Links ]
185. Warshel, A.; Levitt, M. Theoretical studies of enzymic reactions: Dielectric, electrostatic and steric stabilization of the carbonium ion in the reaction of lysozyme. J. Molec. Biol. 1976, 103, 227-249. [ Links ]
186. Dapprich, S.; Komáromi, I.; Byun, K. S.; Morokuma, K.; Frisch, M. J. A new ONIOM implementation in Gaussian98. Part I. The calculation of energies, gradients, vibrational frequencies and electric field derivatives. J. Molec. Struct. THEOCHEM 1999, 461-462, 1-21. [ Links ]
187. Svensson, M.; Humbel, S.; Froese, R. D. J.; Matsubara, T.; Sieber, S.; Morokuma, K. ONIOM: A Multilayered Integrated MO + MM Method for Geometry Optimizations and Single Point Energy Predictions. A Test for Diels−Alder Reactions and Pt(P( t -Bu) 3 ) 2 + H 2 Oxidative Addition. J. Phys. Chem. 1996, 100, 19357-19363. [ Links ]
188. Röhrig, U. F.; Frank, I.; Hutter, J.; Laio, A.; VandeVondele, J.; Rothlisberger, U. QM/MM Car-Parrinello Molecular Dynamics Study of the Solvent Effects on the Ground State and on the First Excited Singlet State of Acetone in Water. ChemPhysChem 2003, 4, 1177-1182. [ Links ]
189. Webb, S. P.; Gordon, M. S. Solvation of the Menshutkin Reaction: A Rigorous Test of the Effective Fragment Method. J. Phys. Chem. A 1999, 103, 1265-1273. [ Links ]
190. Warshel, A.; Weiss, R. M. An empirical valence bond approach for comparing reactions in solutions and in enzymes. J. Am. Chem. Soc. 1980, 102, 6218-6226. [ Links ]
191. Spiegel, K.; Rothlisberger, U.; Carloni, P. Cisplatin Binding to DNA Oligomers from Hybrid Car-Parrinello/Molecular Dynamics Simulations. J. Phys. Chem. B 2004, 108, 2699-2707. [ Links ]
192. Spiegel, K.; Magistrato, A. Modeling anticancer drug-DNA interactions via mixed QM/MM molecular dynamics simulations. Org. Biomolec. Chem. 2006, 4, 2507. [ Links ]
193. Magistrato, A.; Ruggerone, P.; Spiegel, K.; Carloni, P.; Reedijk, J. Binding of Novel Azole-Bridged Dinuclear Platinum(II) Anticancer Drugs to DNA: Insights from Hybrid QM/MM Molecular Dynamics Simulations. J. Phys. Chem. B 2005, 110, 3604-3613. [ Links ]
194. Futera, Z.; Platts, J. A.; Burda, J. V. Binding of piano-stool Ru(II) complexes to DNA; QM/MM study. J. Computat. Chem. 2012, 33, 2092-101. [ Links ]

