










Services on Demand
Journal
Article
 English (pdf)
English (pdf)
 Article in xml format
Article in xml format Article references
Article references
 Send this article by e-mail
Send this article by e-mailIndicators
-
 Cited by SciELO
Cited by SciELO -
 Access statistics
Access statistics
Related links
-
 Similars in
SciELO
Similars in
SciELO
Share

 Permalink
PermalinkJournal of the Mexican Chemical Society
Print version ISSN 1870-249X
J. Mex. Chem. Soc vol.57 n.3 Ciudad de México Jul./Sep. 2013
Article
Spectroscopic Studies and DFT Calculations of Cimetidine Complexes with Transition Metal Ions
Daniela Olea-Román,1 Juan Carlos Villeda-García,2 Raúl Colorado-Peralta,1,4 Alejandro Solano-Peralta,2 Mario Sanchez,3 Irán F. Hernández-Ahuactzi,3 and Silvia Elena Castillo-Blum,1,*
1 Departamento de Química Inorgánica.
2 Unidad de Servicios y Apoyo a la Investigación, Facultad de Química, Universidad Nacional Autónoma de México, Ciudad Universitaria, C.P. 04510, D.F., México.
3 Centro de Investigación en Materiales Avanzados, S.C. Alianza Norte 202, PIIT, Carretera Monterrey-Aeropuerto Km. 10, C.P. 66600, Apodaca N.L., México.
4 Facultad de Ciencias Químicas, Universidad Veracruzana, Prolongación de Oriente 6, No. 1009, Col. Rafael Alvarado, C.P. 94340, Orizaba, Ver., México. blum@unam.mx
Received February 27, 2013
Accepted July 19, 2013
Abstract
The coordination behavior of the antiulcer drug cimetidine (cime) towards transition metal ions was investigated. The synthesis and characterization of [Cr(cime)2Cl2]Cl·3H2O, [Co(cime)Cl2]·5H2O, [Co(cime)3Cl]Cl·3H2O, [Ni(cime)Cl2(H2O)2]·H2O, [Cu(cime)Cl2]·2H2 O, [Cu(cime)2Cl(H2O)]Cl·H2O, [Cu(cime)3Cl2]·3H2O, [Cu2(cime)Cl4], and [Zn(cime)Cl2]·1.5H2O are discussed, where cime acts as monodentate (imidazole N3) or bidentate ligand (N3 and S8). IR, UV-vis, EPR and NMR spectroscopies, mass spectrometry (FAB+), were employed for the characterization. In order to identify the most reactive areas of cimetidine, the electrostatic potential map of the ligand was calculated; also the structures of minimum energy of the coordination compounds were modeled using DFT (B3LYP/def2-TZVP) calculations.
Key words: cimetidine, coordination compounds, spectroscopic characterization, transition metals, DFT calculations, structural analysis.
Resumen
Se investigó la coordinación del antiulcerante cimetidina (cime) frente a iones metálicos de transición. Se discute la síntesis y caracterización de [Cr(cime)2Cl2]Cl·3H2O, [Co(cime)Cl2]·5H2O, [Co(cime)3Cl]Cl·3H2O, [Ni(cime)Cl2(H2O)2]·H2O, [Cu(cime)Cl2]·2H2 O, [Cu(cime)2C l(H2O)]Cl·H2O, [Cu(cime)3Cl2]·3H2O, [Cu2(cime)Cl4], y [Zn(cime)Cl2]·1.5H2O donde cime actúa como ligante monodentado (N3 del imidazol) o bidentado (N3 y S8). Los compuestos se caracterizaron mediante IR, espectroscopía electrónica de absorción, RPE, RMN, espectrometría de masas (FAB+). Con objeto de identificar las áreas más reactivas de la cimetidina, se calculó el potencial electrostático del ligante, así como las estructuras de mínima energía de los compuestos de coordinación, mediante cálculos DFT (B3LYP/def2-TZVP).
Palabras clave: cimetidina, compuestos de coordinación, caracterización espectroscópica, metales de transición, cálculos DFT, análisis estructural.
Introduction
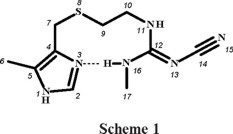
Cimetidine, (scheme 1), 2-cyano-1-methyl-3-(2-[(5-methyl-1H-imidazol-4-yl)-methyl-thio]-ethyl)-guanidine, is a potent histamine H2-receptor antagonist, which inhibits excessive acid secretion caused by histamine, and is used for treatment of peptic ulcer [1]. This drug has the ability to chelate metal ions in blood plasma and in different tissues [2], and it had previously been suggested that the main therapeutic action of cimetidine might be mediated by its interactions with essential metal ions [3]. It resulted interesting to find out whether cimetidine could compete for CuII ion against biological ligands such as albumin [1]. Therefore, the coordination chemistry of cimetidine (cime) has been investigated for some time. The X-ray crystal structures of [Cu(cime)2](ClO4)2, was reported in 1980 [4], later on, the polymeric structure of [Cu(cime)2](NO3)2 was discussed [5] and that of [Cu(cime)2]SO4·9H2O was also published [6]. The X-ray diffraction studies of [Cu(cime)2]X2 (X = ClO4 -, NO3 -, SO42-) reveal solid state structures composed of polymeric cationic CuII complexes and anionic groups [4-6]. The copper ions are six-coordinated, by two imidazole nitrogens and two thioeter sulfur atoms from two different cimetidine molecules; the coordination sphere is completed by two cyano nitrogens from neighbouring molecules. Soto et al [7] and Bianucci [8] synthesised and characterised by IR and UV-vis absorption spectroscopies, coordination compounds of the type [M(cime)2]X2 (M = Co2+, Ni2+, X = NO3 -, BF4 -[7] and ClO4 -[8]). Vibrational spectra (IR and Raman) of cimetidine and their complexes [M(cime)2](ClO4)2, where M corresponds to CuII, CdII, CoII and NiII, were obtained and calculated using semiempirical methods: MNDO, AM1 and PM3 [9].
Electrochemical and potentiometric studies of cimetidine CuI/II complexes were carried out to find out the stability of the complexes in comparison with those formed by other biological ligands; however, no structural discussion is included [1]. In that paper it was shown that the stability of the CuI-cimetidine complex is enormous therefore it can survive in the presence of biological ligands. Also the chemistry of platinum and palladium with cimetidine has been of interest, a potentiometric and spectroscopic study (1H NMR) studies of coordination compounds with Pd and Pt is found in the literature [10, 11] and later on, the crystal structure of trans-[Pt(cime)2]Cl2·12H2O. To determine the antitumor activity of the drugs, the interaction of the metallic complexes and free cimetidine with DNA was assessed [12].
A solventless synthetic procedure was also employed to obtain [Co(cime)2](SO4) and [Ni(cime)2](OAc)2 where the compounds were characterised by spectroscopic and analytical techniques [13]. There is one report in the literature where the stability constants of the ML2 species (M = MnII and NiII) and L = cimetidine [2] were determined by a potentiometric method, where the characterisation of the complexes included IR and 1H NMR spectroscopies. Later on, the stability constant of the [Ni(cime)2]2+ cation in ethanol, at two temperatures, was determined by a spectrophotometric method, showing that a very stable species is formed [14]. The antiulcer activity of a zinc-cimetidine complex in rats has also been studied; however the characterisation of the compound is not discussed [15].
Herein we discuss the synthesis of cimetidine coordination compounds with CrIII, CoII, NiII, CuII and ZnII, and were characterized by several spectroscopic techniques (IR, UV-Vis, 1H and 13C NMR, and EPR), so as by mass spectrometry, and elemental analyses. DFT (B3LYP/def2-TZVP) calculations were carried out in order to know the most reactive areas of cimetidine, as well as the structures of minimum energy of the coordination compounds obtained.
Results and Discussion
There are four known conformation polymorphs of cimetidine: A, B, C and D [16 - 25], that have been characterised by means of IR and 13C NMR spectroscopies [16, 23]. Polymorph A was used in this work, since this is the preferred species used as antiulcer drug [26].
Cimetidine (cime) was reacted with the chlorides of CrIII, CoII, NiII, CuII and ZnII in ethanol, using different molar ratios and reaction conditions, depending upon the metal ion. It was observed that cimetidine coordinated either through the imidazolic nitrogen and the thioether sulfur atom, yielding 5-membered rings, or as monodentate using only the imidazolic nitrogen atom, in all cases (see scheme 2 and scheme 3). The coordination compounds obtained in this work are shown in scheme 2 and scheme 3. A number of different complexes were isolated, since different stoichiometric ratios were employed, all reactions were carried out in ethanol.
Nine coordination compounds (1 -9) were obtained using chlorides of the first row transition metal ions.
Spectroscopic characterization
IR and Electronic Absorption Spectra
Table 1 shows the frequencies of the characteristic vibrations of cimetidine and its coordination compounds. The IR spectrum of cimetidine shows the following stretching vibrations ν(N-H)arom at 3224, ν(C≡N) at 2176, ν(C=N)imd at 1587 and ν(C-S) at 686 cm -1. The spectra of all complexes show the ν(C=N)imd vibration, shifted to higher energies for most complexes, with the exception of [Cr(cime)2(Cl)2]Cl·3H2O (1), [Co(cime)Cl2]·5H2O (2) and [Co(cime)3Cl]Cl·3H2O (3) where a shift towards lower energy was observed. These observations are indicative of coordination through the imidazolic nitrogen atom. The ν(C-S) vibration was shifted to higher energy for most complexes, except for [Co(cime)3Cl]Cl·3H2O 3 and [Cu(cime)3Cl2]·3H2O 7, indicating coordination also through the thioeter sulfur atom for compounds 1, 2, 4 -6, 8 and 9.
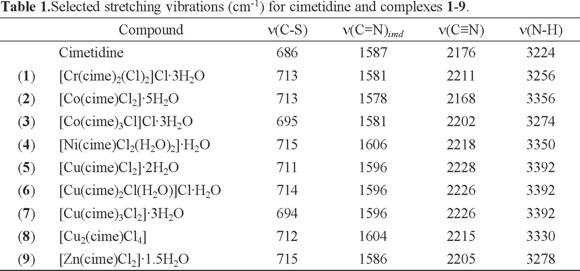
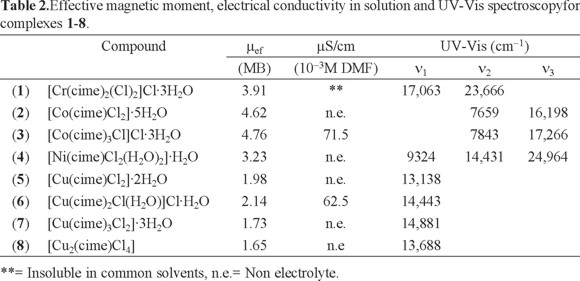
The electronic absorption spectrum (diffuse reflectance) of [Cr(cime)2(Cl)2]Cl·3H2O (1) shows two bands assigned to two of the three spin allowed transitions for octahedral CrIII compounds 4T2g ← 4A2g and 4T1g(F) ← 4A2g at 17063 and 23666 cm-1, while those of [Co(cime)Cl2]·5H2O (2) and [Co(cime)3Cl]Cl·3H2O (3) also display two bands, corresponding to the 4T1(F) ← 4A2 and 4T1(P) ← 4A2 transitions for tetrahedral CoII (ν2 and ν3) at 7659 and 16198, and 7843 and 17266 cm-1, respectively (see Table 2). The spectrum of [Ni(cime)Cl2(H2O)2]·H2O (4) shows three bands at 9324, 14431 and 24964 cm-1 assigned to the 3T2g ← 3A2g, 3T1g (F) ← 3A2g and 3T1g (P) ← 3A2g transitions for octahedral NiII. For the CuII compounds, the electronic absorption spectra display only one d-d band in the region from 13188 to 14881 cm-1, for [Cu(cime)Cl2]·2H2O (5) and [Cu(cime)Cl2]·2H2O (8) it is assigned to the 2E ← 2T transition, while for [Cu(cime)2Cl(H2O)]Cl·H2O (6) to 2T2g ← 2Eg and [Cu(cime)3Cl2]·3H2O (7) where a broad band is observed.
NMR Data
Table 3 shows the 1H and 13C and 1H-1H COSY NMR spectra were recorded in DMSO-d6. When comparing the proton signals of the cimetidine spectrum with those in the spectrum of complex [Zn(cime)Cl2]·1.5H2O (9), it is observed that they are shifted towards higher frequencies when the ligand is coordinated to ZnII, especially those assigned to H-1 (s), that is found at 11.86 ppm for the ligand, and at 12.88 ppm in the complex, H-2 (s) is shifted from 7.47 to 7.95 ppm and H-7 from 3.60 to 3.81, indicating that the ligand is coordinated through the imidazolic nitrogen and the thioeter S atom.
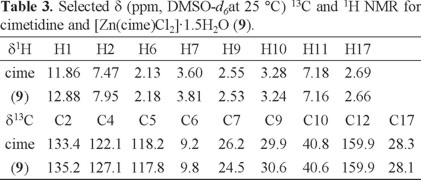
13C signals were assigned using 1H-13C HETCOR spectra. Except for C-4, the signals of the carbon atoms are displayed in the spectra of cimetidine and its zinc complex. The signals for C-2, C-5, C-6 and C-9 were shifted towards higher frequencies, when comparing to those of the free ligand. The largest shifts were for carbon atoms close to N3 and S8. The signal corresponding to C-14 was not observed.
EPR Spectra
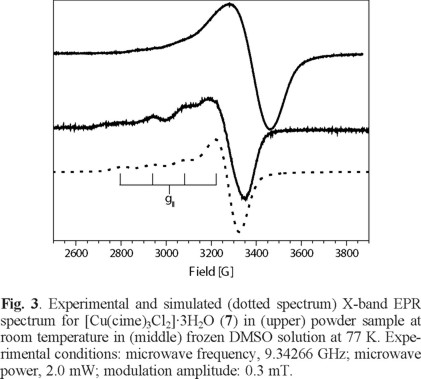
For [Cr(cime)2(Cl)2]Cl⋅3H2O (1), in frozen DMSO solution at 77 K, an axially distorted EPR spectrum is obtained. The g value of 1.98 is typical for CrIII compounds [27].
Several complexes of CuII are reported in the literature, obtained under different reaction conditions [1]. In addition, the structure of the copper cimetidine complex is likely to be an important factor for its biological activity [28]. For example, the antitumor activity of the monomeric CuII aspirin complex ([Cu(Asp)2(Py)2]) is reportedly more effective than the dimeric [Cu2(Asp)4] complex [29]. The magnetic properties of Cu allow, therefore, the application of a range of analytical techniques to assist in the characterization of the coordination around the CuII center. Thus, here the EPR spectra of the mononuclear CuII complexes [Cu(cime)(Cl)2]·2H2O (5), [Cu(cime)2Cl(H2O)]Cl·H2O (6) and [Cu(cime)3Cl2]·3H2O (7) are discussed. For complex (5), the X-band EPR spectrum of a powder sample obtained at room temperature shows a rhombic signal with the following g- values; g1= 2.232, g2= 2.116 y g3= 2.019 with gaver - value of 2.122, see Table 4. This complex in frozen DMSO solution at 77 K shows an axial -type EPR spectrum with g⊥ = 2.086 and g|| = 2.344 (gaver - value of 2.172) and hyperfine splitting in the parallel component with a|| Cu = 142.1 G, see Fig. 1. This change in the gaver - value suggests modification of the compound geometry from distorted tetrahedral in the solid state, while in solution an octahedral environment is favored [30].
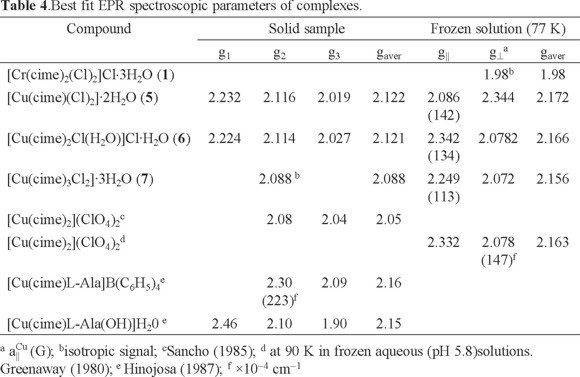
S.-Garcia and coworkers [31] studied the stability of the copper(II) ions complexed with cimetidine in aqueous solutions observing a sharp increase in the absorbance in 2 min, afterwards it remained constant for at least 1 h. This observation indicates changes in the environment around the transition metal ion in solution. Additionally, this spectrum is similar to that reported by Hinojosa [32] of the [Cu(cime)L-Ala(OH)(H20)] complex where a dimeric nature for the latter compound was proposed. In our case, no characteristic signal of dimeric species around 1700 G is observed.
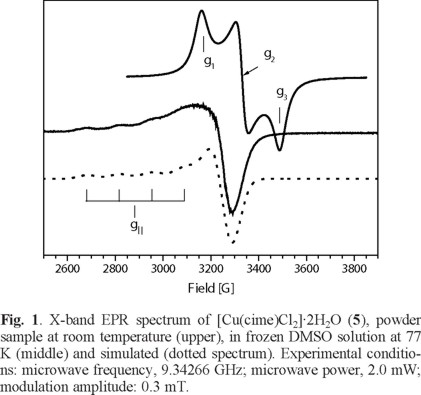
On the other hand, for [Cu(cime)2Cl(H2O)]Cl·H2O (6), the EPR spectrum at room temperature of a powder sample, see Fig. 2, shows a rhombic signal with g-values; g1 = 2.224, g2 =2.114, g3 = 2.027 with a gaver-value of 2.121 corresponding to a CuII in a distorted octahedral geometry [30]. In frozen (0.1 M, DMF) solution at 77 K an axial signal was obtained with the following g-values; g|| = 2.342 and g⊥ = 2.078 with hyperfine (a|| Cu = 134 G) splitting. the gaver - value of 2.166 different from that obtained for the powder sample, this again suggests changes in the environment around the CuII metal center (Table 4).
In this case, Sancho and coworkers [33] reported the EPR spectrum for [Cu(cime)2](ClO4)2 in the solid state, according to the g-values observed, a CuII center in an elongated tetragonal octahedral environment is suggested. However, both Sancho and Greenaway [4] found by X-ray analysis that this complex is formed by an infinite cationic polymer and the perchlorate ions are linked by N-H•••O, hydrogen bonds.
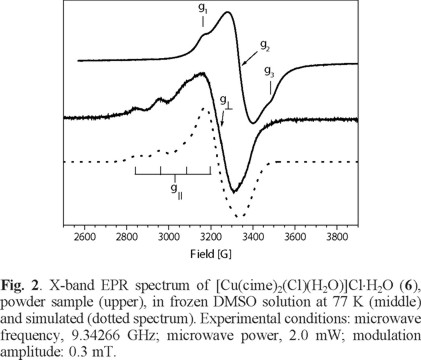
For complex (7), from a powder sample, its EPR spectrum obtained at room temperature shows a pseudo-isotropic signal with g- value of 2.088 while in a frozen DMF solution at 77 K shows a EPR spectrum an axially-distorted signal with gaver - value of 2.156 (g|| = 2.249 and g⊥ = 2.072) and hyperfine splitting in the parallel component (a|| Cu = 113 G), see Fig. 3 and Table 4. These type of EPR spectra are typical of distorted octahedral CuII compounds for the solution sample, where DMF must be coordinated to the Cu centre [28].
Magnetic susceptibility and electrical conductivity
The effective magnetic moments observed for compounds 1 to 8 are within the expected values for CrIII (1), CoII (2, 3), NiII (4) and CuII (5 -8), with three, two and one unpaired electrons. Their molar conductivities measured in DMF are characteristic for 1:1 (3 and 6) electrolytes (see Table 2).
Structural analyses by DFT calculations
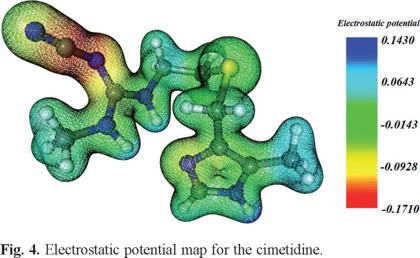
Electrostatic potential maps of three-dimensional molecular diagrams are very useful. They enable us to visualize the charge distribution in the molecules and predict their properties and reactivity; and also allow us to picture the size and shape of molecules.
We calculated the electron density and electrostatic potential map of cimetidine in order to identify its most reactive areas, (see Fig. 4), the red areas are rich in electron density, while those in blue show the areas with low electron density. Although the cyano group is rich in electron density is not coordinating and all hydrogen atoms that are attached to the nitrogen atoms occur on blue zones, that is in electron deficient zones, therefore it was inferred that the coordinating sites of the molecule are the imidazolic nitrogen and the thioeter sulfur. This is consistent with the results presented here in and other reports [4-6, 10-12].
An important aim of this work is the structural analysis of the different complexes and the stabilization evaluation of the different geometries and point groups. The structures of minimum energy were determined by calculating their vibrational modes; all stationary points are true minimal. The calculated results agree with those from the IR and UV-Vis spectroscopic data of the herein synthesized compounds.
The analysis of all possible geometries for compounds 1 -9, by changing the orientation of the bidentate ligands to find out the global minimum, resulted in various symmetries: C1 for 2, 4, 5, 6, 8 and 9; C2 for 1 and C3 for 3 and 7. These were the most stable symmetries for the coordination compounds. Figure 5 and Figure 6 show the most stable structures calculated by the B3LYP/def2-TZVP method. Tetra- and hexacoordinated compounds may stabilize either distorted tetrahedral or octahedral geometries, respectively.
The lowest energy optimized structures of complexes 1 and 6 (C2 and C1 symmetries, respectively), show a preference for inter- ligand interaction of two ligands in the same molecule, between the nitrogen atom of the cyano group and the hydrogen atom of imidazole NH, where the sum of the van der Waals radii N•••H (2.8 Å) are smaller than those calculated using literature data [34], (see Fig. 5). This interaction stabilizes the "U" intra- ligand arrangement for C1 symmetry compounds 2, 5, 8 and 9, see Figure 6. These interactions may be of outmost importance in determining the geometry of the complexes.
According to the calculations trans isomer is the most stable for compounds 1 and 4: [Cr(cime)2Cl2]Cl·3H2O, [Ni(cime)Cl2(H2O)2]·H2O while for 6, [Cu(cime)2Cl(H2O)]Cl·H2O, the cis isomer results more stable than the trans. For compound 7, [Cu(cime)3Cl2]·3H2O, the DFT calculations indicate that its geometry is TBP.
The geometries of copper(II) complexes is either distorted octahedral, distorted tetrahedral or distorted TBP, as expected.
Concluding Remarks
Cimetidine coordinated in most cases as a bidentate ligand, except when three ligands coordinated to the metal ion, where coordination only through the imidazolic nitrogen atom was observed. Four different copper(II) compounds were isolated when the reactions were carried out at different molar ratios, yielding tetrahedral and octahedral complexes. EPR spectra and DFT calculations support the results presented herein.
This contribution includes the synthesis and characterisation of cimetidine complexes with the chlorides of chromium(III), cobalt(II), nickel(II), copper(II) and zinc(II), where spectroscopic and analytical data allow us to propose different structures for the compounds. These proposed structures are in agreement with the spectroscopic and MS data.
Cimetidine acts as bidentate ligand, since hydrogen bonds are formed between the cyano group of a coordinated molecule with the uncoordinated NH of the imidazole, belonging to a second coordinated cimetidine molecule.
Antiulcer activity of the copper(II) compounds is to be tested in a near future.
Experimental
Materials
Metal salts and solvents were purchased from Aldrich, Fluka or Baker (absolute ethanol) and used without further purification.
Cimetidine (was kindly provided by ENCB-IPN, México), Elemental analysis calculated for C10H16N6S, C 47.60, H 6.39, N 33.30, S 12.71; Found C 47.64, H 6.41, N 33.68, S 12.88%. IR, (ν, cm -1) (N-H) (arom) 3224, (C-NH) 3143, (C-NH-CH3) 3036, (CH3) 2920, (C≡N) 2176, (C=C-) 1621, (C=N) 1587, (CH3) 1386, (C-S-) 686. NMR (δ ppm, DMSO-d6) 1H: H1 (s, 11.86), H2 (s, 7.47), H6 (s, 2.13), H7 (s, 3.60), H9 (t, 2.55), H10 (dd, 3.28), H11 (s, 7.18), H16 (s, 7.30) H17 (d, 2.69); 13C NMR (δ ppm, DMSO-d6) C2 133.4, C4 122.1, C5 118.2, C6 9.2, C7 26.2, C9 29.9, C10 40.8, C12 159.9, C17 28.3.
Physical Methods
IR spectra were recorded on KBr pellets on a Perkin Elmer FTIR model 1605 spectrometer from 4000 to 400 cm-1. Elemental Analyses were obtained on a Fisons Instruments model EA1108 analyses, using sulphanilamide as standard. Diffuse reflectance UV-Vis-NIR electronic absorption spectra were determined on a Varian Cary 5E spectrometer, from 40000 to 4000 cm-1. A Johnson Mathey MSB model MK II magnetic balance was used to determine the magnetic susceptibility of the samples. 1H, 13C, 1H-1H COSY and 1H-13C HETCOR NMR spectra were obtained on a Varian Unity Inova (400 MHz) in DMSO-d6.
The EPR spectra were obtained on Brucker Elexsys E-500 spectrometer, both in solid state at room temperature as in frozen DMSO and DMF solution at 77 K using conventional finger dewar. EPR spectra were evaluated and simulated using the Bruker software, and g-values were calculated by measuring accurately the magnetic field and the microwave frequency. Electrical conductivity was determined from 10-3M DMSO solutions using an ORION 140 conductimeter.
Synthesis
[Cr(cime)2(Cl)2]Cl·3H2O, (1). 200 mg of granular Zn0 were treated with conc. HCl and transferred to a filter thimble of a Soxhlet extractor together with 5g of cimetidine and 758 mg of anhydrous CrCl3. 40 mL of methanol were added to the reservoir flask and the methanol was refluxed for 30 minutes. The chromium chloride and cimetidine were extracted and after evaporating the solvent a green paste was obtained. This was washed with ethanol and a green precipitate was isolated, yield 38 % (807.7 mg). Elemental analysis calculated for C20H38O3N12S2Cl3Cr, C 33.50, H 5.34, N 23.44, S 8.94; Found C 33.62, H 5.51, N 21.89, S 8.33%.
[Co(cime)Cl2]·5H2O, (2). 0.5 mmol (118.9 mg) of CoCl2·6H2O and 0.5 mmol (126.1 mg) of cimetidine, were separately dissolved in ethanol (15mL), then mixed and refluxed for 24 h, yielding a blue solution which was concentrated and left for two days yielding a dark blue precipitate, yield 35% (82.7 mg). Elemental analysis calculated for C10H26O5N6SCo, C 25.43, H 5.54, N 17.79, S 6.79; Found C 25.53, H 4.68, N 17.04, S 7.62%. EM (FAB+), (m/z, fragment) 346 [Co(cime)Cl]+, 155 [Co(cime)]2+.
[Co(cime)3Cl]Cl·3H2O, (3). 0.5 mmol (118.9 mg) of CoCl2·6H2O and 1.5 mmol (378.4 mg) of cimetidine, were separately dissolved in ethanol (15mL), then mixed and refluxed for 24 h, yielding a blue solution which was left to stand for two days and a blue solid was isolated, yield 27% (127.0 mg). Elemental analysis calculated for C30H54O3N18S3Cl2Co, C 38.30, H 5.78, N 26.79, S 10.22; Found C 38.72, H 5.56, N 26.11, S 11.04%. EM (FAB+), (m/z, fragment) 850 [Co(cime)3Cl]+, 598 [Co(cime)2Cl]+, 346 [Co(cime)Cl]+, 155 [Co(cime)]2+.
[Ni(cime)Cl2(H2O)2]·H2O, (4). 118 mg (0.5 mmol) of NiCl2 ·6H2O was dissolved in 15 mL of ethanol, and mixed with a solution of 126.1 mg (0.5 mmol) of cimetidine in 15 mL of ethanol. The mixture was stirred for 24 h and a light green precipitate was isolated and washed with ethanol, yield 51% (111.1 mg). Elemental analysis calculated for C10H22O3N6SCl2Ni, C 27.65, H 5.07, N 19.35, S 7.37; Found C 27.67, H 5.28, N 18.55, S 6.91%. EM (FAB+), (m/z, fragment) 345 [Ni(cime)Cl]+, 155 [Ni(cime)]2+.
[Cu(cime)Cl2]·2H2O, (5). 84.4 mg (0.5 mmol) of CuCl2 ·2H2O were dissolved in 15 mL of etanol, and mixed with 15 mL of a cimetidine ethanolic solution (126.1 mg, 0.5 mmol). The mixture was stirred for 24 h and a green precipitate was isolated and washed with ethanol, yield 85% (179.7 mg). Elemental analysis calculated for C10H20O2N6SCl2Cu, C 28.50, H 4.75, N 19.95, S 7.60; Found C 28.91, H 4.68, N 19.75, S 8.14%. EM (FAB+), (m/z, fragment) 350 [Cu(cime)Cl]+, 158 [Cu(cime)]2+.
[Cu(cime)2Cl(H2O)]Cl·H2O, (6). 84.4 mg (0.5 mmol) of CuCl2 ·2H2O were dissolved in 15 mL of ethanol, and mixed with 15 mL of a cimetidine ethanolic solution (252.3 mg, 1.0 mmol). The mixture was stirred for 24 h and a green precipitate was isolated and washed with ethanol, yield 79% (266.6 mg). Elemental analysis calculated for C20H36O2N12S2Cl2Cu, C 35.66, H 5.35, N 24.96, S 9.51; Found C 34.97, H 5.10, N 24.07, S 9.94%. EM (FAB+), (m/z, fragment) 602 [Cu(cime)2Cl]+, 350 [Cu(cime)Cl]+, 284 [Cu(cime)2]2+,158 [Cu(cime)]2+.
[Cu(cime)3Cl2(H2O)]·2H2O, (7). 84.4 mg (0.5 mmol) of CuCl2 ·2H2O were dissolved in 15 mL of ethanol, and mixed with 15 mL of a cimetidine ethanolic solution (378.4 mg, 1.5 mmol). The mixture was stirred for 24 h and a green precipitate was isolated and washed with etanol, yield 53% (250.6 mg). Elemental analysis calculated for C30H54O3N18S3Cl2Cu, C 38.18, H 5.73, N 26.72, S 10.19; Found C 37.76, H 5.48, N 25.93, S 10.87%. EM (FAB+), (m/z, fragment) 602 [Cu(cime)2Cl]+, 410 [Cu(cime)3]2+, 284 [Cu(cime)2]2+, 158 [Cu(cime)]2+.
[Cu2(cime)Cl4], (8). 168.8 mg (1.0 mmol) of CuCl2 ·2H2O were dissolved in 15 mL of ethanol, and mixed with 15 mL of a cimetidine ethanolic solution (126.1 mg, 0.5 mmol). The mixture was stirred for 24 h and a green precipitate was isolated and washed with ethanol, yield 81% (211.1 mg). Elemental analysis calculated for C10H16N6SCl4Cu2, C 23.04, H 3.1, N 16.12, S 6.15; Found C 23.34, H 4.27, N 15.88, S 7.05%. EM (FAB+), (m/z, fragment) 519 [Cu2(cime)Cl4]+, 483 [Cu2(cime)Cl3]+, 422 [Cu(cime)Cl3]+, 224 [Cu2(cime)Cl2]2+, 138 [Cu2(cime)Cl]3+, 95 [Cu2(cime)]4+.
[Zn(cime)Cl2]·1.5H2O, (9). 136.28 mg (1.0 mmol) of ZnCl2 were dissolved in 15 mL of ethanol, and mixed with 15 mL of a cimetidine ethanolic solution (252.3 mg, 1.0 mmol). A precipitate was isolated and washed with ethanol, yield 89% (369.3 mg). NMR (δ ppm, DMSO-d6) 1H: H1 (s, 12.88), H2 (s, 7.95), H6 (s, 2.18), H7 (s, 3.81), H9 (t, 2.53), H10 (t, 3.24), H11 (s, 7.16), H16 (s, 7.30) H17 (s, 2.66); 13C NMR (δ ppm, DMSO-d6) C2 135.2, C4 127.1, C5 117.8, C6 9.8, C7 24.5, C9 30.6, C10 40.6, C12 159.9, C17 28.1. Elemental analysis calculated for C20H40O8N12S3Zn, C 28.90, H 4.61, N 20.22, S 7.71; Found C 29.75, H 4.57, N 20.78, S 7.83%. EM (FAB+), (m/z, fragment) 351 [Zn(cime)Cl]+, 158 [Zn(cime)]2+.
Computational details
The geometry of all structures was fully optimized using the density functional theory (B3LYP) [35, 37] in combination with the def2-TZVP basis set using the Gaussian 09 software package [38], and their vibrational frequencies were computed at the same level of theory. All calculations are true minimal, since the values of the vibrational frequencies are all positive. Results were visualized by using the Chemcraft v1.6 program [38].
Acknowledgements
We acknowledge financial support from Project IN206707 PAPIIT-DGAPAUNAM and CONACyT-SEP E-43662-F. We thank M. Gutiérrez-Franco, N. López-Balbiaux, V. Lemus-Neri, R. I. del Villar, G. Duarte and R. P. Fierro-Ramírez for technical support.
References
1. Kimura, E.; Koike, T.; Shimizu, Y.; Kodamalb M. Inorg. Chem. 1986, 25, 2242-2246. [ Links ]
2. Kanumfre, F.; de Lima, E. M.; Scheidt, G.; Carneiro, P. I. B.; Rosso, N. D. J. Braz. Chem. Soc. 2010, 21, 800-805. [ Links ]
3. Bonora, S.; Foggia, M. D.; Tugnoli, V.; Righi, V.; Benassic, E.; Maris, A. J. Raman Spectrosc. 2011, 42, 612-620. [ Links ]
4. Greenaway, F. T.; Brown, L. M.; Dabrowiak, J. C.; Thompson, M. R. Day, V. W. J. Am. Chem. Soc. 1980, 102, 7782-7784. [ Links ]
5. Soto, L.; Borrás, J.; Sancho, A.; Fuertes, A.; Miravitlles, C. Acta Cryst. 1985, C41, 1431-1433. [ Links ]
6. Soto, L.; Legros, J. P.Polyhedron 1988, 7, 307-314. [ Links ]
7. Abadia, A.; Sancho, A.; Soto, L.; Borrás, J. Transition Met. Chem. 1986, 11, 8-11. [ Links ]
8. Bianucci, A. M.; Demartin, F.; Manassero, M.; Masciocchi N.; Ganadu, M. L.; Naldini, L.; Panzanelli, A. Inorg. Chim. Acta 1991, 182, 197-204. [ Links ]
9. Barańska, M.; Proniewicz, L. M. J. Mol. Struct. 1999, 511-512, 153-162. [ Links ]
10. Nurchi, V.; Cristiani, F.; Crisponi, G.; Ganadu, M. L.; Lubinu, G.; Panzanelli, A.; Naldini, L. Polyhedron 1992, 11, 2723-2727. [ Links ]
11. Crisponi, G.; Cristiani, F.; Nurchi, V. M.; Silvagni, R.; Ganadu, M. L.; Lubinu, G.; Naldini, L.; Panzanelli, A. Polyhedron 1995, 14, 15171530. [ Links ]
12. Onoa, G. B.; Moreno, V.; Freisinger, E.; Lippert, B. J. Inorg. Biochem. 2002, 89, 237-247. [ Links ]
13. Tella, A. C; Eke, U. B.; Isaac, A. Y.; Ojekanmi, C. A. Orbital Elec. J. Chem,. Campo Grande, 2011, 3, 94-103. [ Links ]
14. Tirmizi, S. A.; Wattoo, F. H.; Wattoo, M. H. S.; Sarwar, S.; Memon, A. N.; Iqbal, J.; Ghanghro, A. B. Arabian J. Chem. 2012, 5, 309-314. [ Links ]
15. Ito, M.; Inaguma, K.; Suzuki, Y.; Segami, T.; Suzuki. Y. Japan. J. Pharmacol, 1995, 68, 287-295. [ Links ]
16. Shibata, M.; Kokubo, H.; Morimoto, K.; Morisaka, K.; Ishida, T.; Inoue, M. J. Pharm. Sci. 1983, 72, 1436-1442. [ Links ]
17. Mitchell, R. C. J. Chem. Soc. Perkin Trans. 2 1980, 915-918. [ Links ]
18. Hädicke, E.; Frickel, F.; Franke, A. Chem. Ber. 1978, 111, 3222-3232. [ Links ]
19. Byrn, S. R.; Graber, C. W.; Midland, S. L. J. Org. Chem. 1976, 41, 2283-2288. [ Links ]
20. Saran, A.; Srivastava, S.; Kulkarni, V. M.; Coutinho, E. Indian J. Biochem. Biophys. 1992, 29, 54-64. [ Links ]
21. Tudor, A. M.; Davies, M. C.; Melia, C. D.; Lee, D. C.; Mitchell, R. C.; Hendra, P. J.; Church, S. J. Spectrochim. Acta Part A: Mol. Spectroscopy 1991, 47, 1389-1393. [ Links ]
22. Kojic-Prodic, P.; Kajfer, F.; Belin, B.; Toso, R.; Sunjic, V. Gazz. Chim. Ital., 1979, 109, 535-539. [ Links ]
23. Middleton, D. A.; Le Duff, C. S.; Berst, F.; Reid, D. G. J. Pharm. Sci. 1997, 86, 1400-1402. [ Links ]
24. Párkányi, L.; Kálmán, A.; Hegedüs, B.; Harsányi, K.; Kreidl, J. Acta Crystallogr. 1984, C40, 676-679. [ Links ]
25. Danesh, A.; Chen, X.; Davies, M. C.; Roberts, C. J.; Sanders, G. H. W.; Tendler, S. J. B.; Williams, P. M.; Wilkins, M. J. Langmuir 2000, 16, 866. [ Links ]
26. United States Pharmacopeial Convention. USP XXIII. United States Pharmacopeia. 23 ed. Easton: Mack Printing; 1995. [ Links ]
27. Kumar, R.; Singh, R. Turk. J. Chem.; 2006, 30, 77- 87. [ Links ]
28. Weder, J. E.; Dillon, C. T.; Hambley, T. W.; Kennedy, B. J.; Lay, P. A.; Biffin, J. R.; Regtop, H. L.; Davies, N. M.; Coord. Chem. Rev. 2002, 232, 95-126. [ Links ]
29. Oberley, L.W.; Buettner, G.R.; Cancer Res. 1979, 39, 1141-1149. [ Links ]
30. El-Ayaan, U.; Gabr. I. M. Spectrochim. Acta Part A 2007, 67, 263-272. [ Links ]
31. García, M. S.; Albero, M. I.; Sánchez-Pedreño, C.; Abuherba, M.S.; J. Pharm. Biomed. Anal. 2003, 32, 1003-1010. [ Links ]
32. Hinojosa, M.; Ortiz, R.; Perelló, L.; Borrás, J.; J. Inorg. Biochem., 1987, 29, 119-129. [ Links ]
33. Sancho, A.; Borras, J.; Soto-Tuero, L.; Esteban-Calderón, C.; Martínez-Ripoll M.; García-Blanco, S.; Polyhedron, 1985, 4, 539-543. [ Links ]
34. Batsanov, S. S. Inorg. Mater. 2001, 37, 871-885. [ Links ]
35. Becke, A. D. J. Chem. Phys. 1993, 98, 5648-5652. [ Links ]
36. Lee, C.; Yang, W.; Parr, R. G. Phys. Rev. 1988, B37, 785-789. [ Links ]
37. Gaussian 09, Revision B.01, Frisch, M. J.; Trucks, G. W.; Schlegel, H. B.; Scuseria, G. E.; Robb, M. A.; Cheeseman, J. R.; Scalmani, G.; Barone, V.; Mennucci, B.; Petersson, G. A.; Nakatsuji, H.; Caricato, M.; Li, X.; Hratchian, H. P.; Izmaylov, A. F.; Bloino, J.; Zheng, G.; Sonnenberg, J. L.; Hada, M.; Ehara, M.; Toyota, K.; Fukuda, R.; Hasegawa, J.; Ishida, M.; Nakajima, T.; Honda, Y.; Kitao, O.; Nakai, H.; Vreven, T.; Montgomery, Jr., J. A.; Peralta, J. E.; Ogliaro, F.; Bearpark, M.; Heyd, J. J.; Brothers, E.; Kudin, K. N.; Staroverov, V. N.; Kobayashi, R.; Normand, J.; Raghavachari, K.; Rendell, A.; Burant, J. C.; Iyengar, S. S.; Tomasi, J.; Cossi, M.; Rega, N.; Millam, J. M.; Klene, M.; Knox, J. E.; Cross, J. B.; Bakken, V.; Adamo, C.; Jaramillo, J.; Gomperts, R.; Stratmann, R. E.; Yazyev, O.; Austin, A. J.; Cammi, R.; Pomelli, C.; Ochterski, J. W.; Martin, R. L.; Morokuma, K.; Zakrzewski, V. G.; Voth, G. A.; Salvador, P.; Dannenberg, J. J.; Dapprich, S.; Daniels, A. D.; Farkas, Ö.; Foresman, J. B.; Ortiz, J. V.; Cioslowski, J.; Fox, D. J. Gaussian, Inc., Wallingford CT, 2009. [ Links ]
38. ChemCraft v1.6, G.A. Zhurko, G. A.; Zhurko, D. A. [ Links ]

