










Servicios Personalizados
Revista
Articulo
 Inglés (pdf)
Inglés (pdf)
 Artículo en XML
Artículo en XML Referencias del artículo
Referencias del artículo
 Enviar artículo por email
Enviar artículo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO -
 Accesos
Accesos
Links relacionados
-
 Similares en
SciELO
Similares en
SciELO
Compartir

 Permalink
PermalinkJournal of the Mexican Chemical Society
versión impresa ISSN 1870-249X
J. Mex. Chem. Soc vol.57 no.3 Ciudad de México jul./sep. 2013
Article
Synthesis and Anticancer Activity of Gold(I)-Chloroquine Complexes
Maribel Navarro,1,3,* William Castro,1 Sorenlis González,1 María Jesús Abad,2 and Peter Taylor2
1 Centro de Química, Instituto Venezolano de Investigaciones Científicas (IVIC). Caracas 1020-A, Venezuela. m.navarroacosta@murdoch.edu.au, maribelnava@gmail.com
2 Centro de Medicinal Experimental, Instituto Venezolano de Investigaciones Científicas, IVIC Caracas 1020-A, Venezuela.
3 Present Address: Chemistry and Analytical Sciences, School of Veterinary and life Sciences, Murdoch University, Western Australia 6150, Australia.
Received February 27, 2013
Accepted June 18, 2013
Abstract
Two new gold(I) -chloroquine complexes, Au(CQ)(Cl) (1) and Au(CQ)(tgta) (2), were prepared and their most probable structure were established through a combination of different spectroscopic and analytical techniques. Their interaction with two important targets of action, DNA and thioredoxin reductase (TrxR), were investigated. These studies showed that complexes 1 and 2 displayed two types of interaction with DNA, covalent binding through the metal center, and additionally a non-covalent interaction that is electrostatic in the case of complex 1, but intercalative for complex 2, which is similar to that displayed by free CQ. The experimental data indicated that these gold-CQ complexes also possess the ability to inhibit TrxR. These results led us to test their cytotoxicity against 6 tumor cell lines. The complexes displayed cytotoxic activity against the PC-3, SKBR-3, HT-29, LoVo and B16/BL6 lines. These finding suggest that gold(I)-CQ compounds, particularly [Au(CQ)(PPh3)]PF6, are promising chemotherapeutic alternatives in the search of anticancer agents.
Key words: Chloroquine, gold, anticancer agents, DNA and thioredoxin reductase.
Resumen
Dos nuevos complejos de oro(I)-cloroquina: Au(CQ)(Cl) (1) y Au(CQ)(tgta) (2) fueron sintetizados, la estructura más probable para cada uno de ellos fue establecida a través de la combinación de diversas técnicas espectroscópicas. Se evaluó la interacción de cada uno de estos nuevos compuestos con dos importantes blancos de acción: ADN y tioredoxina reductasa (TrxR). Dichos estudios indicaron que 1 y 2 son capaces de interaccionar con el ADN a través de dos vía: covalentemente a través del centro metálico, y no covalentemente, en el caso del complejo 1 es del tipo electrostática mientras que para el complejo 2 es del tipo intercalativa. Además, la data experimental indicó que ambos complejos 1 y 2 tienen la capacidad de inhibir el sistema TrxR. Estos resultados motivaron la evaluación de sus actividad citotóxica contra 6 líneas celulares tumorales, encontrándose que, los complejos mostraron actividad citotoxica contra las líneas PC-3, SKBR-3, HT-29, LoVo y B16/BL6. Estos alentadores resultados sugieren que los complejos Au(I)-CQ, particularmente el [Au(CQ)(PPh3)]PF6 son una alternativa prometedora en la búsqueda de agentes anti cancerígenos.
Palabras clave: Cloroquina, oro, agentes anticancerígenos, ADN, tioredoxina reductas.
Introduction
Gold complexes have attracted significant attention as potential anticancer compounds, due to the fact that many gold(I) and gold(III) compounds inhibit the growth of cancer cells, including those in cisplatin-resistant tumors [1-5]. The cytotoxic effects of these complexes are associated with DNA damage and mitochondrial function inhibition, and their interaction with numerous intracellular targets such as nitrogen bases, cysteine, glutathione reductase, selenocysteine and thioredoxin reductase [6-11].
A large percentage of chemotherapeutic anticancer drugs interact with DNA directly or prevent the proper relaxation of DNA (through the inhibition of topoisomerases); these drugs may work at any phase of the cell cycle, but are most effective during DNA synthesis. The interactions between these compounds and DNA have been divided into covalent and non-covalent, with the most representative example of covalent interaction being with platinum complexes, which covalently bind to DNA, forming DNA adducts and activating various signal-transduction pathways, for example those involved in DNA-damage recognition and repair, cell-cycle arrest, and programmed cell death/apoptosis. In the case of non-covalent interactions (groove binders and intercalators), the compounds can mediate their cytotoxic activity by enhancing the ability of the enzymes to generate DNA breaks and/or prevent relegation [12].
Another important target is selenoenzyme thioredoxin reductase (TrxR), which is a NADPH-dependent flavoprotein responsible for a large number of functions in DNA synthesis, defense against oxidative stress and apoptosis or redox signaling, as well as cell homeostasis regulation [13-14]. A mechanism suggested for the inhibition is covalent binding with a selenocysteine residue in the C-terminal active site of TrxR [15]. With respect to cytotoxicity, it has recently been proposed [16-17] that binding of Au(I) at the active site in mitochondrial TrxR promotes apoptosis since H2O2 produced by the mitochondrial respiratory chain oxidizes reducing agents such as thioredoxins which then cannot be reduced by Au(I)-inhibited TrxR [18]. Oxidised TrxR can act on several different membrane targets leading to increased permeability of the mitochondrial membranes, release of cytochrome c and cell death. Specific targeting to tumor cells may depend on the elevated mitochondrial membrane potential of cancer cells. Studies on potential anticancer gold compounds are presently being performed to understand the cellular and molecular mechanisms through which these compounds induce their important antiproliferative effects.
Worthy of mention is chloroquine (CQ), which has been shown to display anticancer [19-20] as well as cancer protective activities [21-22], and also to enhance the potency of other drugs [23-24]. In a previous study, we demonstrated that complexing of palladium [25] or platinum [26] to CQ increased their cytotoxicity compared to the free ligand. These results encouraged us to continue evaluating promising clinical metals such as gold(I), and lead us to present here the synthesis and full characterization of two new Au(I)-CQ complexes. Their interaction with two important targets of action, DNA and thioredoxin reductase (TrxR), were investigated through various physical and spectroscopic techniques and in terms of their cytotoxic activity against tumor cell lines.
Experiments
Reagent
All manipulations were routinely carried out under N2 using common Schlenk techniques. Solvents were purified by standard procedures immediately prior to use. CQDP, calf thymus DNA (CT-DNA), buffers and solvents were purchased from Sigma-Aldrich Co. The extraction of the CQ base [27], and the synthesis of Au(THT)Cl (THT = tetrahydrothiophene) [28] have been described previously. All other commercial reagents were used without further purification. The NMR spectra were obtained in a DMSO-d6 solution in a Bruker AVANCE 300 spectrometer. 1H and 13C NMR shifts were recorded relative to residual proton resonances in the deuterated solvent. IR spectra were obtained with a Thermo Scientific Nicolet instrument. Ultraviolet-visible (UV-vis) spectra were recorded on a HP 8453 diode array instrument. Electrospray ionization mass spectrometry (ESI-MS) spectra were obtained using a Thermo Finnigan LXQ with methanol as the solvent. Conductivity measurements were performed with a LaMotte CDS 5000 conductimeter. The [Au(CQ)(PPh3)]PF6 complex was prepared according to the literature [29]
Synthesis of new complexes
Au(CQ)(Cl) [1]
A solution of Au(THT)Cl (144.7 mg, 0.45 mmol) in dichloromethane (10 mL) was stirred until complete dissolution was achieved, then a solution of CQ (148.0 mg, 0.46 mmol) was added. The stirring was continued for 2 h at -60 °C. The volume of the solvent was reduced under a nitrogen stream, and diethyl ether was added until the solution became turbid. On cooling of the sample to -10 °C overnight, a white product precipitated which was filtered, washed with diethyl ether, and dried under vacuum. Yield 60%. Elemental analysis (%) Calc. for C18H26N3Cl2Au: C 39.1; N 7.6; H 4.8. Found: C 39.2; N 7.5; H 4.6. ESI-MS (CH2Cl2) [M+H]+: 552.21; IR: υ (N-H) 3358 cm-1; υ (C=C) 1612 cm-1; υ (C=N) 1549 cm-1. UV-vis (DMSO) 262 and 349 nm. ε (DMSO) [(λ nm)]: 17200 M-1 cm -1 (262 nm) and 16200 M-1 cm -1 (349 nm). 1H NMR (DMSO-d6; δ ppm): 8.53 (1H; d; J, 1.95 Hz); 8.38 (1H; d; J, 6.60 Hz); 7.86 (1H; d; J, 8.94 Hz); 7.24 (1H; dd; J1, 1.95 Hz; J2, 8.94 Hz); 7.11 (1H; d; J, 6.27 Hz); 6.41(1H; d; J, 6.69 Hz); 3.36 (1H; m); 2.10 (6H, m); 1.78 (4H; m); 1.22 (3H; d; J, 6.21 Hz); 1.18 (6H, t); 13C-NMR (DMSO-d6; δ ppm): 157.27; 150.23;146.69; 135.23; 126.35; 126.85; 122.37; 117.48; 98.75; 52.36; 48.34; 46.90; 33.10; 22.83; 19.89; 10.03. Molar conductivity in DMSO, ΛM = 12 ± 5 ohm-1 cm2 mol-1.
Au(CQ)(tgta) [2]
A solution of Au(THT)Cl (101.2 mg, 0.32 mmol) in dichloromethane (10 mL) was stirred until complete dissolution was achieved, then a solution of CQ (108.9 mg, 0.34 mmol) was added and stirred for 2 h at -60 °C. After adding 1-thio-β-D-glucose tetraacetate (tgta) (120.24 mg, 0.33 mmol) in dichloromethane, the mixture was stirred at -60 °C for 2 h. The volume of the solvent was reduced under a nitrogen stream, and diethyl ether was added until the solution became turbid. On cooling of the sample to -10 °C overnight, a white product precipitated which was filtered, washed with diethyl ether, and dried under vacuum. Yield 59%. Elemental analysis (%) Calc. for C32H45N3ClO9SAu C 43.7; N 4.8; H 5.2; S 3.6 Found: C 43.4; N 5.0; H 5.0; S 3.4. ESI-MS (CH2Cl2) [M+H]+: 880.47; IR: υ (N-H) 3467 cm-1; υ (CO-O) 1744 cm-1; υ (C=C) 1620 cm-1; υ (C=N) 1574 cm-1; υ (S-C) 601 cm -1. UV-vis (DMSO) 256 and 343 nm. ε (DMSO) [(λ nm)]: 14600 M-1 cm -1 (256 nm) and 14200 M-1 cm -1 (343 nm). 1H NMR (DMSO-d6; δ ppm): 8.56 (1H, d, J = 9.12); 8.44 (1H, d, J = 6.24 Hz); 7.94 (1H, d, J = 7.86 Hz); 7.86 (1H, d, J = 1.96 Hz); 7.57 (1H, dd, J1 = 9.03 Hz, J2 = 2.10 Hz,); 6.70 (1H, d, J = 6.39 Hz); 5.16 (1H, m); 4.87 (1H, t); 4.72 (1H, m); 4.09 (2H, dd); 3.02 (2H, m); 2.34 (6H, m); 1.60 (4H, m); 1.25 (3H, d, J = 6.00 Hz); 1.96 (12H);1.14 (6H, t); 13C-NMR (DMSO-d6; δ): 210,61; 207.9; 203.72; 201.6; 156.72; 151.65; 146.91; 136.89;125.73; 124.62; 122.02; 117.88; 99.03; 70.98; 70.49; 54.23; 52.28; 49.67; 47.06; 47,73; 46.43; 44.65; 33.83; 29.45; 22.92; 19.77; 10.59. Molar conductivity in DMSO, ΛM = 13 ± 5 ohm-1 cm2 mol-1.
Interaction with biomolecules
DNA interaction studies
For the covalent binding studies, the gold complexes were mixed with CT-DNA and incubated for 72 h (Ri 0.2, 1 mL of metal complex and 1 mL DNA). DNA was precipitated by adding ethanol (2X sample volume) and 2 M NaCl (0.1X sample volume). After centrifugation, the supernatant was removed and the DNA resuspended in water overnight. This precipitation-resuspension cycle was repeated three times and the final suspension was analyzed for Au by ICP atomic emission spectrometry and for DNA with the Burton assay [30].
The spectrophotometric titrations were carried out by stepwise additions of a CT DNA solution (1 mM, in 5 mM Tris-HCl, pH 7.2and 50 mM NaCl buffer) to a solution of each complex (~70 μM) in DMSO, then recording the UV-vis spectra at 330 and 343 nm after each addition. The absorption of DNA was subtracted by adding the same amounts of CT DNA to the blank. The binding affinities were obtained by using the Scatchard equation, r/Cf = K(n-1), for ligand macromolecule interactions with noncooperative binding sites [31-34], where r is the number of moles of Au complex bound to 1 mol of CT DNA (Cb/CDNA), n is the number of equivalent binding sites, and K is the affinity of the complex for those sites. Concentrations of free (Cf) and bound (Cb) complex 1 were calculated from Cf = C(1-α) and Cb = C-Cf, respectively, where C is the total Au concentration. The fraction of bound complex (α) was calculated according to α = (Af-A)/(Af-Ab), where Af and Ab are the absorbance of the free and fully bound complex at the selected wavelength, and A is the absorbance at any given point during the titration. Kb is obtained from the slope of the plot [35].
To measure the interaction of complex 1 and 2 with CT-DNA by fluorimetric titration, the excitation and emission wavelengths were set to 343 and 380 nm, respectively. Using standard right-angle emission optics, we recorded fluorescence intensity measurements using the photon counting mode and corrected for any fluctuations of the 450-W xenon arc lamp source by deflecting a portion of the excitation signal onto a separate photodiode. The fluorimetric titration was carried out at room temperature. The complex was dissolved in a buffer consisting of 70% DMSO and 30% Tris-HCl (5 mM Tris-HCl and 50 mM NaCl; pH 7.4) to obtain a 700 μM solution. Twenty μL of that stock solution were then diluted with 1980 μL of the same buffer in a quartz cuvette and then titrated with 10 μL additions of a 90 μM solution of CT DNA (5 mM Tris-HCl (pH 7.2), 50 mM NaCl). Emission spectra were monitored at 380 and 550 nm until saturation was reached. The binding affinities were obtained using the Scatchard equation r/Cf = K(n-1).
Viscosity measurements were carried out using an Ostwald viscometer immersed in a water bath maintained at 25 °C. The DNA concentration (75 μM in 5 mM Tris-HCl (pH 7.2), 50 mM NaCl) was kept constant in all samples, while the complex concentration was increased from 0 to 67 μM. Flow time was measured at least 6 times with a digital stopwatch and the mean value was calculated. Data are presented as (η/η0)1/3 versus the ratio [complex]/[DNA], where η and η0 are the specific viscosities of the DNA in the presence and absence of the complex, respectively. The values of η and η0 were calculated from the expression (t -tb)/tb, where t is the observed flow time and tb is the flow time of buffer alone. The relative viscosity of the DNA was calculated from η/η0 [36].
For the circular dichroism measurements, a solution of each complex was freshly prepared in DMSO (5 mM). The appropriate volumes of that solution were added to 3 mL samples of a freshly prepared solution of CT DNA (195 μM) in Tris-HCl buffer (5 mM Tris-HCl, 50 mM NaCl, pH = 7.29) to achieve molar ratios of 0-0.5 drug/DNA. The samples were incubated at 37 °C for 18 h. All CD spectra of DNA and of the DNA-drug adducts were recorded at 25 °C over the range 220-330 nm and finally corrected with a blank and using noise reduction. The final data is expressed in molar ellipticity (millidegrees) [37].
For the DNA electrophoresis assays, 10 μL samples of the plasmid pBR322 (20 μg/mL) were combined with the complex at different ratios and then incubated for 18 h at 37 °C. Five μL of each sample were run (100 mV for 45 min) on a 1% agarose gel with TBE-1X (0.45 M Tris-HCl, 0.45 M boric acid, 10 mM EDTA) and stained with ethidium bromide (5 μL ethidium bromide per 50 mL agarose gel mixture). The bands were then viewed with a trans-luminator and the image captured with a camera [38].
Inhibition of thioredoxin reductase (TrxR) activity
Rat liver TrxR (Sigma) was used to determine TrxR inhibition by the compounds. The assay was performed according to the manufacturer's instructions (Sigma product information sheet T9698). The compounds were freshly dissolved as stock solutions in DMSO. In a 96-well plate, 100 µL of reaction mixture was added to each well. This mixture contained, per ml, 567 µl of distilled water, 400 µl of 500 mM potassium phosphate buffer, pH 7.0, containing 50 mM EDTA; 20 µL of BSA solution 20 mg/mL, 10 µl of NADPH 40 mg/mL and 3 µL of thiorredoxin reductase. Two µL of the compounds in graded concentrations or DMSO (control probe), were added and were incubated for 10 min at room temperature. The reaction was started with the addition of 98 µl of 20 mM DTNB solution in DMSO. After mixing, the formation of TNB was monitored in a microplate reader (TECAN sunrise absorbance reader) at 405 nm at 15 s intervals for 16 min. The increase in TNB concentration over time followed a linear trend (r2 ≥ 0.99), and the enzymatic activities were calculated as the slopes (increase of absorbance per second) thereof. For each tested compound, the non-interference with the essay components was confirmed by a negative control experiment using an enzyme-free solution. The IC50 values were calculated as the concentration of compound decreasing the enzymatic activity of the untreated control by 50% and are given as the mean and error of repeated experiments [39].
Growth inhibition and cytotoxicity testing
Five human and one murine tumor cell lines were used. HT-29 and LoVo (human colon carcinoma), MCF-7 and SKBR-3 (human breast carcinoma), PC-3 (human prostate carcinoma) and B16/BL6 (murine melanoma) cells were cultured in Dulbecco's Modified Eagle's Medium (DMEM) supplemented with 10% heat-inactivated fetal bovine, (Gibco, BRL, USA) and penicillin (100 Units/mL) - streptomycin (100 μg/mL), containing in addition glucose 0.45% for the HT-29 cells. The sulphorhodamine B (SRB) assay was used to evaluate the effect of the compounds on the growth and viability of the six tumor cell lines [40]. Each drug was assayed in triplicate at 6 different concentrations up to a maximum of 30 µM. The concentrations inducing 50% growth inhibition (GI50), total growth inhibition (TGI) and 50% cytotoxicity (LC50) after a 48 h incubation period were calculated by linear interpolation from the observed data points.
Results and discussion
Synthesis and characterization
Au(CQ)(Cl)(1)
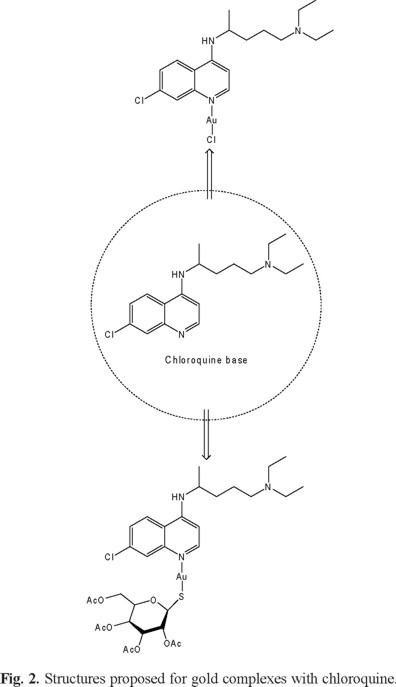
Reaction of Au(THT)(Cl) (THT = tetrahydrothiophene) with CQ at low temperature (-60 °C) for 2 h leads to the new complex Au(CQ)(Cl) (1), which was isolated in good yield (60%). Elemental analyses of this complex are in agreement with the molecular formula proposed. The IR spectra of the complexes displayed peaks clearly associated with the presence of the coordinated CQ (Displacement of peaks with respect to free CQ: 15 cm-1 (N-H), 5 cm-1 (C=C), 9 cm-1 (C=N)). The ESI-MS spectrum of complex 1 displayed parent peaks of high intensity corresponding to its molecular ion (M + H) at m/z 552.21. This feature confirms its molecular weight and that the fragmentation pathways were initiated by the loss of chloride ion giving (M-Cl) at m/z 516.24 (Fig. 1). The new complex was well characterized by NMR spectroscopy; all resonances could be unequivocally assigned on the basis of 1D and 2D correlated COSY and HMQC experiments. The 1H and 31C chemical shift variation of each signal with respect to those of the free ligand (Δδ) was used as a parameter to deduce the mode of bonding of CQ to the metal. It has been previously shown by us [25-26] and by others [41] that the largest variations are always observed for the protons and carbons located in the vicinity of the N-atom attached to the metal. Selected data contained in Table 1 indicate that CQ binds to the metal in complex 1 through the quinoline ring N(1) atom, which is a good donor site of this molecule. Correspondingly, the largest shift with respect to the free ligand is observed for H(8) (∆δ 0.89), H(5) (∆δ 0.43), H(2) (∆δ 0.39) and H(6) (∆δ 0.29), located in the vicinity of N(1) while all other chloroquine protons shift by <0.30 ppm except NH which moves 1.79 ppm downfield. Similarly, in 31C NMR, the carbons with the largest shift with respect to the free ligand were observed for C2, C9 and C8 (∆δ 7.31, 6.50 and 4.29 respectively). The molar conductivity values obtained for complex 1 are in the range corresponding to neutral complexes dissolved in DMSO [42]. From this data, we postulate the structure shown in figure 2A, the formulation for 1 thus corresponding to the usual 14electron linear configuration for Au(I).
Au(CQ)(tgta)(2)
The synthesis of the new complex 2, Au(CQ)(tgta) (TgTa= 1-thio-β-D-glucose tetraacetate), was also carried out at low temperature (-60 °C) by the reaction of Au(THT)(Cl), with CQ and TgTa, in good yield (59%) as a stable white powder. Elemental analyses of complex 2 are in agreement with the molecular formula proposed. The ESI-MS spectrum for complex 2 displays the molecular ion peak (M-H) at m/z 880.47 with high intensity (Fig. 3). The IR spectra of the complex displayed peaks clearly associated with the presence of the coordinated of both ligand (CQ and TgTa) (Displacement of peaks with respect to free CQ: 18 cm-1 (N-H), 9 cm-1 (C=C), 4 cm-1 (C=N)). On the basis of the data shown in Table 1, we propose that CQ is bound to the metal through the N1 atom of the quinoline moiety, a good donor site of this molecule, since large shifts with respect to free CQ were observed for H8, H5 and H2 (Δδ = 0.22, 0.27 and 0.45 respectively), C2 (∆δ = 6.76), C9 (∆δ = 5.05) and C8 (∆δ = 4.88), while all other protons and carbon signals suffered minor displacements with respect to free CQ. On the other hand, in the case of the tgta, large shifts with respect to free TgTa were observed for Ha and Hg (∆δ = 0.22 and 0.8); Ca and Cg (∆δ = 6.12 and 7.4), suggesting that the ligand binds to the metal through the sulfur atom. These findings suggest that complex 2 is non-electrolytic, which was verified by the corresponding molar conductivity values, which were below the expected range for electrolyte 1:1 [42]. Based on the analysis of the results we proposed the structure shown in figure 2B, since the formulation for complex 2 corresponds to a 14-electron Au(I) complex linear coordination geometry.
Though no X-ray determination is available for both complexes, the structures could be established through a combination of physical and spectroscopic analyses, primarily NMR, providing convincing support for the proposed structures.
Interaction with biomolecules
DNA interaction studies
Studies of the interaction of anticancer gold complexes with DNA indicate firstly, that gold(I) and gold(III) complexes may interact with DNA via different chemical mechanisms to produce different conformational changes in the DNA, and secondly, that certain coordinating ligands in gold complexes (e.g. Cl, Br and SCN) can be exchanged for binding sites on DNA by gold [43]. All these non-covalent interaction with DNA suggest that the antitumor activity of many gold complexes is not the result of direct interaction with DNA and many researchers propose that mitochondria and pathways of oxidative phosphorylation may be their primary intracellular targets [44]. However, DNA remains an important therapeutic target for diseases such as cancer. Moreover, chloroquine has been shown to bind to DNA through intercalation and electrostatic interactions and this mode of binding is retained in some metal-CQ complexes through the coordinated CQ moiety, although it is not clear if such DNA binding is responsible for their antitumor activity [45-46]. Therefore, it was important to establish whether the new Au-CQ derivatives bind effectively to DNA, and determine what types of interactions are playing the major roles, in order to get an idea of their potential as possible antitumor agents.
The results of covalent binding studies for complexes 1 and 2 shown in Table 2 indicate that while 1 binds at around 2.35 Au atoms/base, 2 is bound at 1.03 Au atoms/base. These binding levels are greater than those observed for others metal complexes with known anticancer activity such as cisplatin (0.74, Table 2), and others compounds that present important activity, such as platinum [47] and rhodium complexes [48]. This may be taken as evidence that covalent Au-DNA binding is taking place with complexes 1 and 2. The levels of covalent binding of cisplatin and transplatin with DNA, as measured by us and also included in Table 2 for comparison, are in agreement with those previously reported values [47] and are lower than our values for complexes 1 and 2.

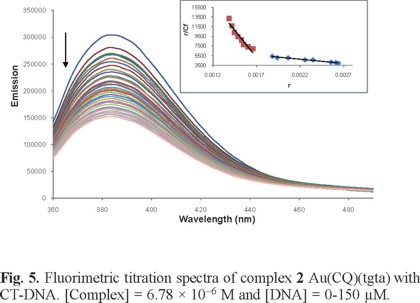
In order to shed further light on the interactions of complexes 1 and 2 with DNA, we performed absorption and emission titration experiments. The absorption plots showed that adding DNA to solutions of each complex to saturation caused hypochromism at the absorbance maxima (330 and 345 nm) and an isosbestic point at 297 nm. As an example, the data for complex 1 are shown in Fig. 4 and the corresponding binding constants (Kb) for all the complexes are summarized in Table 3. The values for complexes 1 and 2 are very similar to those for CQ, lying within the interval for which a compound is considered to be interacting with DNA [49-50]. They are also comparable to Kb values obtained for other transition metal-(CQ) complexes [26, 51-52]. The emission bands of complexes 1 and 2 at 390 nm decreased in intensity (Figure 5) as DNA was added to saturation. The binding constants calculated from a Scatchard plot for the data at the emission maxima are shown in Table 3. These values, obtained for CQ and other metal-CQ complexes, indicated that the complexes interact with DNA in a manner analogous to free CQ and are consistent with those ones reported elsewhere for other metal complexes [53]. Such interactions have been described in terms of reversible processes such as intercalation through the planar CQ moiety [54], although other reversible interactions such as hydrogen bridging or electrostatic binding may be taking place as well, in addition to the covalent interaction proposed above.

CD spectroscopy has been widely used to examine changes in DNA morphology during drug-DNA interactions, as the band due to base stacking (275 nm) and that due to right-handed helicity (248 nm) are quite sensitive to the mode of interaction with small molecules [55]. Figure 6 shows the spectra of all complexes evaluated with CT DNA solutions at different ratios, as well as the CD spectrum of DNA alone. Complex 1 decreased the ellipticity of the positive band and caused changes in the negative band, while complex 2 increased the positive band but produced no significant changes in the ellipticity of the negative band. These alterations (Figure 6) are indicative of important modifications by complexes 1 and 2 in the DNA tertiary structure [56].
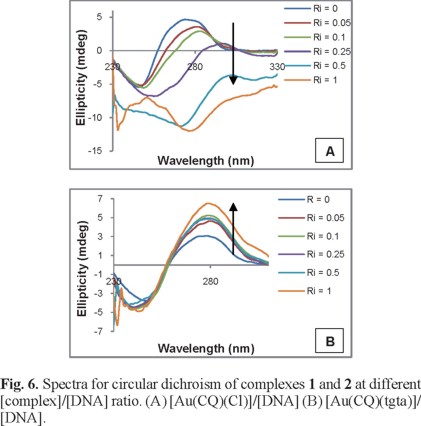
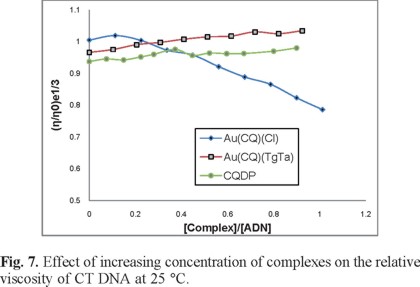
Figure 7 shows the results of a comparative viscosity study designed to explore the mode of binding of the gold compounds to DNA. Hydrodynamic data provide perhaps the most critical test for intercalative binding in the absence of X-ray crystallographic or NMR structural data [57]. Complex 2 cause a significant increase in the viscosity of a DNA solution, which is typical of intercalating compounds such as the classic intercalator ethidium as well as other metallo-compounds [58-59], due to an increase in the separation of the base pairs at the intercalation site and hence an increase in the overall DNA molecular length. On the other hand, complex 1 caused a decrease in the relative viscosity, suggesting a non-intercalative binding mode. Such behavior may be explained by a covalent binding mode that produces bends or kinks in the DNA helix [60-61].
Circular plasmid DNA studies are ideally suited to probe cleavage events, as DNA exists in a supercoiled state in its native form and converts to a relaxed form upon single strand cleavage, exhibiting an altered migration rate in agarose gel electrophoresis [62-63]. Figure 8 shows the plasmid in the presence of different molar ratios of complexes 1 and 2. Line 2 displays the difference in mobility of the plasmid alone (control line), line 3 corresponds to the plasmid incubated with cisplatin, while lines 4-6 correspond to the plasmid incubated with different concentrations of the complexes. The increase in the concentration of complex 1 caused changes in the mobility of the plasmid. For complex 1 at Ri 0.5, only one band is observed, attributable to the circular form, while at higher Ri, no bands are visible for DNA, either relaxed or linear, which we suggest may be due to an intense interaction of the complex with the plasmid. In contrast, complex 2 produced no change in plasmid mobility, suggesting that it did not cause cleavage of the DNA supercoil.
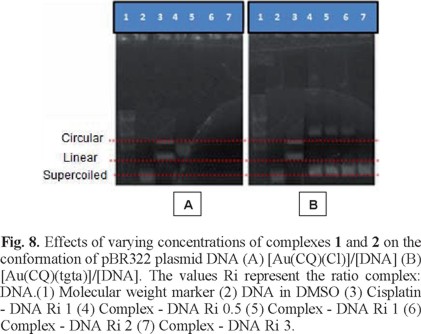
Putting together all the results of the gold-chloroquine -DNA experimental data, we propose that complexes 1 and 2 displayed two types of interaction with DNA, the first showing covalent binding through the metal center, with an additional non-covalent interaction, which, from the data, appears to be electrostatic, while complex 2 appears to intercalate, in a manner similar to that displayed by free CQ. The interaction of complex 3 with DNA has been evaluated in a previous study [64], showing mainly intercalation and electrostatic association associated with the CQ moiety. However, no covalent metal-DNA binding was observed. The interactions that take place between these gold-CQ complexes and the DNA helix may be important in increasing the biological activity of the Au(I)-CQ complexes relative to the free ligand.
Inhibition of thioredoxin reductase (TrxR) activity
The literature reports a number of studies on gold complexes with TrxR inhibitory properties showing the relevance of this enzyme in the pharmacology of gold-based pharmaceuticals [65-67]. Due to its antioxidant properties, the thioredoxin system prevents cells from oxidative stress, which is a key factor in DNA damage. Overexpression of TrxR has been reported in numerous tumor cell lines. Moreover, high levels of the substrate thioredoxin have been associated with resistance to cisplatin. Hence TrxR is considered an attractive target for the development of novel anticancer agents [68]. It has been observed that apoptosis induction by auranofin in Jurkat T cells appears to be mediated by inhibition of the cytosolic and mitochondrial forms of TrxR and was accompanied by an increase in cellular hydrogen peroxide levels. The inhibitory potential of complexes 1 and 2, [Au(CQ)(PPh3)]PF6 (complex 3) and Et3PAuCl (as the positive control), were studied using the dithiobisnitrobenzoic acid (DTNB) reduction assay using isolated rat liver TrxR. This assay makes use of the fact that TrxR reduces the disulfide bonds of DTNB with formation of 2-nitro-5-thionitrobenzoic acid (TNB), which can be detected photometrically.
All gold complexes (1-3) displayed activity against TrxR (Table 4), being even more active than Et3PAuCl, and other gold complexes reported in the literature [69-71]. As expected, the inhibitory effect depends on the presence of a gold center, CQ alone being completely inactive.

The inhibition of TrxR by these complexes suggests that this mitochondrial protein is a potential cellular target, and that disruption of mitochondrial function may be a potential mechanism of tumor cell death. Hence, as the complexes both interact with DNA and inhibit TrxR, they represent strong candidates for evaluation of their anticancer activity.
Growth inhibition and cytotoxicity testing
The compounds were tested on five human tumor cell lines, representing tumors of three different origins, prostate, breast and colon, in addition to a murine melanoma line, which we regularly use for in vivo testing of anticancer drugs that show promising results in vitro. The results are shown in Table 5. As many drugs show a cytostatic effect at doses appreciably lower than those causing cytotoxicity, we used the SRB assay which has the advantage over tetrazolium assays of being able to distinguish between cytostasis (cell growth inhibition) and cytotoxicity (cell death).
Complexes 1 and 2 both exerted some degree of growth inhibition on four of the tumor cell lines and total growth inhibition on the HT-29 and LoVo lines at concentrations below 30 µM. Their general activity was greater than that shown by CQ and cisplatin control compounds, but not as marked as the lead compound 3 [29, 64], which was included here for comparison. The low activity of cisplatin seen in these assays, when compared to its well-known cytotoxicity as reported in the literature, is probably due to the relatively short incubation times used here (48 h). Thus, although we have favorable results for the cytostatic activity of the complexes when compared to cisplatin, further experiments must be performed to determine whether the complexes would show cytotoxic activity, perhaps at longer incubation times, or using more specific assays for cell death.
Conclusion
The synthesis and characterization by of two new gold-chloroquine (1 and 2) complexes were achieved. These complexes interact with DNA through both covalent binding and non-covalent interactions such as electrostatic contacts and/or hydrogen bonding, and intercalation. They both inhibit TrxR. Complex 3 exerted growth inhibition on the human tumor cell lines, showing promise as potential anticancer drugs deserving of further investigation.
Acknowledgments
This work was partially funded by Grant MC 2007000881 from the "Misión Ciencia" - Venezuela. Authors want to thank Prof. Roberto Sánchez Delgado for his assistance with the CD experiments and Prof. Germán Fraile for his suggestions and criticisms. W.C. is grateful to FONACIT for a visiting fellowship.
References
1. Chi-Ming, C.; Wai-Yin, S. R. Chem. Comm. 2011, 47, 9554-9560. [ Links ]
2. Gandin, V.; Fernandes, A. P.; Rigobello, M. P.; Dani, B.; Sorrentino, F.; Tisato, F.; Bjornstedt, M.; Bindoli, A.; Sturaro, A.; Rella, R.; Marzano, C. Biochem. Pharm. 2010, 79, 90-101. [ Links ]
3. Milacic, V.; Ping Dou, Q. Coord. Chem. Rev. 2009, 253, 1649-1660. [ Links ]
4. Lessa, J. A.; Guerra, J. C.; De Miranda, L. F.; Romeiro, C.F.D.; Da Silva, J.G.; Mendes, I. C.; Speziali, N. L.; Souza-Fagundes, E. M.; Beraldo, H. J. Inorg. Biochem. 2011, 105 1729-1739. [ Links ]
5. Barnard, P. J.; Berners-Price, S. J. Coord. Chem. Rev. 2007, 251, 1889-1902. [ Links ]
6. Arner, E. S. J.; Holmgren, A. Semin. Cancer Biol. 2006, 16, 420-426. [ Links ]
7. Zeng, H.H.; Wang, L.H. Med. Chem. 2010, 6, 286-297. [ Links ]
8. Rigobello, M. P.; Messori, L.; Marcon, G.; Cinellu, M. A.; Bragadin, M.; Folda, A.; Scutari, G.; Bindoli, A. J. Inorg. Biochem. 2004, 98, 1634-1641. [ Links ]
9. Arnér, E. S. J. Biochim. Biophys. Acta 2009, 1790, 495-526. [ Links ]
10. Berners-Price, S. J.; Filipovska, A. Australian J. Chem. 2008, 61, 661-668. [ Links ]
11. Messori, L.; Marcon, G.; Orioli, P. Bioinorg. Chem. Applic. 2003, 1, 177-187. [ Links ]
12. Froelich-Ammon, S.J.; Osheroff, N. J. Biol. Chem. 1995, 270, 21429-21432. [ Links ]
13. Kaakoush, N. O.; Mendz, G. L. Curr. Enzy. Inhib. 2009, 5, 117-133 [ Links ]
14. Krishnamurthy, D.; Karver, M. R.; Fiorillo, E.; Orrú, V.; Stanford, S. M.; Bottini, N.; Barrios, A. M. J. Med. Chem. 2008, 51, 4790-4795 [ Links ]
15. Rubbiani, R.; Can, S.; Kitanovic, I.; Alborzinia, H.; Stefanopoulou, M.; Kokoschka, M.; Mönchgesang, S.; Sheldrick, W. S.; Wölfl, S.; Ott, I. J. Med. Chem. 2011, 54, 8646-8657. [ Links ]
16. Rigobello, M.P.; Folda, A.; Dani, B.; Menabo, R.: Scutari, G.; Bindoli, A. Eur. J. Pharmacol. 2008, 582, 26-34. [ Links ]
17. Marzano, C.; Gandin, V.; Folda, A.; Scutari, G.; Bindoli, A.; Rigobello, M.P. Free Rad. Biol. Med. 2007, 42, 872-881. [ Links ]
18. Nordberg, J.; Arner, E. S. J. Free Radic. Biol. Med. 2001, 31, 1287-1312 [ Links ]
19. Sasaki, K.; Tsuno, N.H.; Sunami, E.; Tsurita, G.; Kawai, K.; Okaji, Y.; Nishikawa, T.; Shuno, Y.; Hongo, K.; Hiyoshi, M.; Kaneko, M.; Kitayama, J.; Takahashi, K.; Nagawa, H. BMC Cancer 2010, 10, 370-381. [ Links ]
20. Kimura, T.; Takabatake, Y.; Takahashi, A.; Isaka, Y. Cancer Res. 2013, 73; 3 [ Links ]
21. Maclean, K. H.; Dorsey, F. C.; Cleveland, J. L.; Kastan, M. B. J. Clin. Invest. 2008, 118, 79-88. [ Links ]
22. Dang, C. V. J. Clin. Invest. 2008, 118, 15-17. [ Links ]
23. Zamora, J. M.; Pearce, H. L.; Beck, W. T. Mol. Pharmacol. 1988, 33, 454-462. [ Links ]
24. Sorensen, M.; Sehested, M.; Jensen, P. B. Biochem. Pharmacol. 1997, 54, 373-380. [ Links ]
25. Navarro, M.; Prieto Peña, N.; Colmenares, I.; González, T.; Arsenak, M.; Taylor P.; J. Inorg. Biochem. 2006, 100, 152-157. [ Links ]
26. Navarro, M.; Castro, W.; Higuera-Padilla, A. R.; Sierraalta, A.; Abad, M. J.; Taylor, P.; Sánchez-Delgado, R. A. J. Inorg. Biochem. 2011, 105, 1684-1691. [ Links ]
27. Sánchez-Delgado, R. A.; Navarro, M.; Pérez H.; Urbina, J. A. J. Med. Chem. 1996, 39, 1095-1099. [ Links ]
28. Uson, R.; Laguna, A.; Laguna, M.; Briggs, D. A.; Murray, H. H.; Fackler, J. P. Inorg. Synth. 1989, 26, 85-91. [ Links ]
29. Navarro, M.; Pérez, H.; Sánchez-Delgado, R.A. J. Med. Chem. 1997, 40, 1937-1939. [ Links ]
30. Burton, K. Biochemistry J. 1956, 162, 315-323. [ Links ]
31. McGhee, J.D.; Von Hippel, P.H. J. Mol, Biol. 1974, 86, 469-489. [ Links ]
32. Wei, C.; Jia, G.; Yuan, J.; Feng, Z.; Li, C. Biochemistry 2006, 45, 6681-6691. [ Links ]
33. Boyer, R.F. Biochemistry Laboratory: Modern Theory and Techniques, Benjamin Cummings, San Francisco, 2006. [ Links ]
34. Cusumano, M.; Di Pietro, M.L.; Giannetto, A. Inorg. Chem. 1999, 38, 1754-1758. [ Links ]
35. Satyanarayana, S.; Dabrowiak, J.C. Biochemistry 1992, 31, 9319-9324. [ Links ]
36. Haq, I.; Lincoln, P.; Suh, D.; Norden, B.; Chowdhry, B.Z.; Chaires, J.B. J. Am. Chem. Soc. 1995, 117, 4788-4796. [ Links ]
37. Macquet, J.P.; Butour, J.L. Eur. J. Biochem. 1978, 83, 375-387. [ Links ]
38. Zhang, B.; Seki, S.; Akiyama, K.; Tsutsui, K.; Li, T.; Nagao, K. Acta Med. Okayama. 1992, 46, 427-434. [ Links ]
39. Lessa, J. A.; Guerra, J. C.; de Miranda, L. F.; Romeiro, C. F.D.; Da Silva J. G., Mendes, I. C.; Speziali, N. L.; Souza-Fagundes, E. M.; Beraldo, H. J. Inorg. Biochem. 2011, 105, 1729-1739. [ Links ]
40. Skehan, P.; Storeng, R.; Scudiero, D.; Monks, A.; McMahon, J.; Vistica, D.; Warren, J. T.; Bokesch, H.; Kenney, S.; Boyd, M. R. J. Natl. Cancer Inst. 1990, 82, 1107-1112. [ Links ]
41. Sundquist, W. I.; Bancroft, D. P.; Lippard, S. J. J. Am. Chem. Soc. 1990, 112, 1590-1596. [ Links ]
42. Geary, W. Coord. Chem. Rev. 1971, 7, 81-122. [ Links ]
43. Mirabelli, C. K.; Sung, C. M.; Zimmerman, J. P.; Hill, D. T.; Mong, S.; Crooke, S. T. Biochem. Pharmacol. 1986, 35, 1427-1433. [ Links ]
44. Barnard, P. J.; Berners-Price, S. J. Coord. Chem. Rev. 2007, 251, 1889-1902. [ Links ]
45. Fan, C.; Wang, W.; Zhao, B.; Zhang, S.; Miao, J. Bioorg. Med. Chem. 2006, 14, 3218-3222. [ Links ]
46. Martirosyan, A. R.; Rahim-Bata, R.; Freeman, A. B.; Clarke, C. D.; Howard, R. L.; Strobl, J. S. Biochem. Pharmacol. 2004, 68, 17291738. [ Links ]
47. Navarro, M.; Castro, W.; Higuera-Padilla, A. R.; Sierraalta, A.; Abad, M. J.; Taylor, P.; Sánchez-Delgado, R. A. J. Inorg. Biochem. 2011, 105, 1684-1691. [ Links ]
48. Mahnken, R. E.; Billadeau, M. A.; Nikonowicz, E. P.; Morrison, H. J. Am. Chem. Soc. 1992, 114, 9253- 9265. [ Links ]
49. Long, E. C.; Barton, J. K. Acc. Chem. Res. 1990, 23, 271-273. [ Links ]
50. Graves, D. E.; Watkins, C. L.; Yielding, L. W. Biochem. 1981, 20, 1887-1892. [ Links ]
51. Martínez, A.; Rajapakse, C. S. K.; Jalloh, D.; Dautriche, C.; Sánchez-Delgado, R. A. J. Biol. Inorg. Chem. 2009, 14, 863-871. [ Links ]
52. Navarro, M.; Castro, W.; Higuera-Padilla, A.R.; Sierraalta, A.; Abad, M.J.; Taylor, P.; Sánchez-Delgado R.A. J. Inorg. Biochem. 2011, 105, 1684-1691. [ Links ]
53. Li, Y.; Yang, Z.Y. Inorg. Chim. Acta 2009, 362, 4823-4831. [ Links ]
54. Krajewski, W. A. FEBS Lett. 1995, 361, 149-152. [ Links ]
55. Ivanov, V.I.; Minchenkova, L.E.; Schyolkina, A.K.; Poletayer, A.I. Biopolymers 1973, 12, 89-110. [ Links ]
56. Zhang, B.; Seki, S.; Akiyama, K.; Tsutsui, K.; Li, T.; Nagao, K. Acta Med. Okayama 1992, 46, 427-434. [ Links ]
57. Satyanarayana, S.; Dabrowiak, J. C. Biochemistry 1992, 31, 9319-9324. [ Links ]
58. Cusumano, M.; Di Pietro, M. L.; Giannetto, A. Inorg.Chem. 1999, 38, 1754-1758. [ Links ]
59. Brodie, C. R.; Collins, J. G.; Aldrich-Wright, J. R. Dalton Trans. 2004, 1145-1152. [ Links ]
60. Kapicak, L.; Gabbay, E. J. J. Am. Chem. Soc. 1975, 97, 403-408. [ Links ]
61. Navarro, M.; Betancourt, A.; Hernández, C.; Marchán E. J. Braz. Chem. Soc. 2008, 19, 1355-1360 [ Links ]
62. Holder, A.A.; Swavey, S.; Brewer, K.J. Inorg. Chem. 2004, 43, 303-308. [ Links ]
63. Jin, Y.; Lewis, M.A.; Gokhale, N.H.; Long, E.C.; Cowan, J.A. J. Am. Chem. Soc. 2007, 129, 8353-8361. [ Links ]
64. Navarro, M.; Castro, W.; Martínez A.; Sánchez Delgado R. A. J. Inorg. Biochem. 2011, 105, 276-282. [ Links ]
65. Berners-Price, S. J.; Filipovska, A. Metallomics 2011, 3, 863-873. [ Links ]
66. Nobili, S.; Mini, E.; Landini, I.; Gabbiani, C.; Casini, A.; Messori, L. Med. Res. Rev. 2010, 30, 550-580. [ Links ]
67. Rubbiani, R.; Kitanovic, I.; Alborzinia, H.; Can, S.; Kitanovic, A.; Onambele, L. A. ; Stefanopoulou, M.; Geldmacher, Y.; Sheldrick, W. S.; Wolber, G.; Prokop, A.; Wölfl, S.; Ott, I. J. Med. Chem. 2010, 53, 8608-8618. [ Links ]
68. Bindoli, A., Rigobello, M.P., Scutari, G.; Gabbiani, C.; Casini, A.; Messori, L. Coord. Chem. Rev. 2009, 253, 1692-1707. [ Links ]
69. Barnard, P. J.; Berners-Price, S. J. Coord. Chem. Rev. 2007, 251, 1889-1902. [ Links ]
70. Lessa, J. A.; Guerra, J. C.; de Miranda, L. F.; Romeiro, C. F.D.; Da Silva, J. G.; Mendes, I. C.; Speziali, N. L.; Souza-Fagundes, E. M.; Beraldo, H. J. Inorg. Biochem. 2011, 105, 1729-1739. [ Links ]
71. Liu, W.; Bensdorf, K.; Proetto, M.; Abram, U.; Hagenbach, A.; Gust, R. J. Med. Chem. 2011, 54, 8605-8615. [ Links ]

