










Services on Demand
Journal
Article
 English (pdf)
English (pdf)
 Article in xml format
Article in xml format Article references
Article references
 Send this article by e-mail
Send this article by e-mailIndicators
-
 Cited by SciELO
Cited by SciELO -
 Access statistics
Access statistics
Related links
-
 Similars in
SciELO
Similars in
SciELO
Share

 Permalink
PermalinkJournal of the Mexican Chemical Society
Print version ISSN 1870-249X
J. Mex. Chem. Soc vol.57 n.3 Ciudad de México Jul./Sep. 2013
Article
Spectroscopic Study of the Interactions of Ruthenium-Ketoconazole Complexes of Known Antiparasitic Activity with Human Serum Albumin and Apotransferrin
Jesús G. Estrada and Roberto A. Sánchez-Delgado*
Brooklyn College and The Graduate Center, The City University of New York 2900 Bedford Ave., Brooklyn, NY 11210, USA.
Received June 17, 2013
Accepted July 19, 2013
Abstract
The pharmacological properties of any drug are related to its ability to interact with macromolecular blood components. The interaction of human serum albumin (HSA) and apotransferrin (ATf) with six RuII complexes containing ketoconazole (KTZ), which we have previously reported to be active against Leishmania major and Trypanosoma cruzi, has been investigated by monitoring the tryptophan fluorescence intensity of each protein upon incremental addition of the complexes. All the Ru-KTZ derivatives, namely cis-fac-[RuIICl2(DMSO)3(KTZ)] (1), cis-[RuIICl2(bipy)(DMSO)(KTZ)] (2), [RuII(η6-p-cymene)Cl2(KTZ)] (3), [RuII(η6-p-cymene)(en)(KTZ)][BF4]2 (4), [RuII(η6-p-cymene)(bipy)(K TZ)][BF4]2 (5), and [RuII(η6-p-cymene)(acac)(KTZ)][BF4] (6) are able to quench the intrinsic fluorescence of HSA and ATf at 27 ºC. Analysis of the spectroscopic data using Stern-Volmer plots indicates that in both cases the quenching takes place principally through a static mechanism involving the formation of Ru complex-protein adducts; further analysis of the fluorescence data allowed the estimation of apparent association constants and the number of binding sites for each protein and each compound. The results indicate that both HSA and ATf are possible effective transporters for Ru-KTZ antiparasitic drugs.
Keywords: Ruthenium, ketoconazole, antiparasitic, albumin, transferrin, fluorescence.
Resumen
Las propiedades farmacológicas de cualquier droga están relacionadas con su capacidad para interaccionar con componentes macromoleculares de la sangre. En este artículo se describe un estudio de la interacción de la albúmina sérica humana (ASH) y de la apotransferrina (ATf) con seis complejos de RuII con ketoconazol (KTZ), previamente reportados por nosotros como activos contra Leishmania major y Trypanosoma cruzi. La investigación se realizó mediante el seguimiento de la intensidad de la fluorescencia del triptófano de las proteínas, en función de la cantidad añadida de cada complejo. Todos los compuestos Ru-KTZ estudiados, cis-fac-[RuIICl2(DMSO)3(KTZ)] (1), cis-[RuIICl2(bipy)(DMSO)(KTZ)] (2), [RuII(η6-p-cymene)Cl2(KTZ)] (3), [RuII(η6-p-cymene)(en)(KTZ)][BF4]2 (4), [RuII(η6-p-cymene)(bipy)(KTZ)][BF4]2 (5) y [RuII(η6-p-cymene)(acac)(KTZ)][BF4] (6), son capaces de producir el "quenching" o extinción de la fluorescencia intrínseca de la ASH y de la ATf a 27 ºC. El análisis de los datos espectroscópicos, utilizando la ecuación de Stern-Volmer, indica que en ambos casos el "quenching" se debe principalmente a un mecanismo estático, lo que implica la formación de aductos entre las proteínas y los complejos metálicos. El tratamiento de los datos de fluorescencia permitió además estimar las constantes aparentes de asociación y el número de sitios de enlace para cada proteína y cada complejo. Los resultados sugieren que la ASH y la ATf son posibles trasportadores efectivos para estas drogas antiparasitarias derivadas de la unidad Ru-KTZ.
Palabras clave: Rutenio, ketoconazol, antiparasitarios, albumina, transferrina, fluorescencia.
Introduction
Trypanosomatid infections are responsible for some of the most neglected parasitic diseases that afflict the poor populations of developing countries in the tropical regions of the planet. They include Leishmaniasis, caused by a hemoflagellate protozoan of the genus Leishmania, and American Trypanosomiasis (Chagas' disease), caused by Trypanosoma cruzi. Leishmaniasis is endemic in 88 countries where 350 million people are at risk and about 2 million are diagnosed every year [1-3]. Chagas' disease, in turn, causes over 10,000 deaths and affects over 20 million people in Central and South America [4, 5].
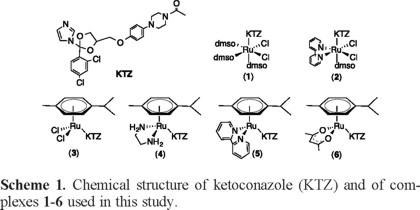
Treatments for these two diseases are scarce, not very effective, and generally toxic, and therefore there is a clear need for new improved therapies [6-9]. For several years we have been searching for metal-based drugs against Chagas and Leishmaniasis, based on the combination of an organic compound of known antiparasitic activity with a metal-containing fragment in a single molecule. Our hypothesis is that the metal can act as a carrier for the organic compound, stabilizing it until it reaches the target, while at the same time the organic compound serves as a carrier for the metal ion in its transit toward a second target, thereby avoiding undesirable metal interactions with other biomolecules. Such metal-drug synergy may result in enhanced activity against parasites and reduced toxicity to human cells, in relation to the individual components of the drug [10, 11]. A number of azole-type molecules, including the well-known antifungal agents clotrimazole (CTZ) and ketoconazole (KTZ), have been recognized as potential agents against trypanosomatid infections due to their ability to inhibit the biosynthesis of sterols that are essential to the parasite.12 We have demonstrated that binding such sterol biosynthesis inhibitors (SBIs) to metal-containing fragments effectively enhances their activity against Leishmania major and Trypanosoma cruzi while displaying low toxicity for mammalian cells [10, 11, 13-15]. Of particular interest to this paper, we recently reported the synthesis of the RuII-KTZ complexes depicted in Scheme 1, cis-fac-[RuIICl2(DMSO)3(KTZ)] (1), cis-[RuIICl2(bipy)(DMSO)(KTZ)] (2), [RuII(η6-p-cymene)Cl2(KTZ)] (3), [RuII(η6-pcymene)(en)(KTZ)][BF4]2 (4), [RuII(η6-p-cymene)(bipy)(KTZ)][BF4]2 (5), and [RuII(η6-p-cymene)(acac)(KTZ)][BF4] (6), and their activity against L. major and T. cruzi [16].
As part of our ongoing investigations on the biological potential of these metal-based drugs, we are exploring possible transport mechanisms. The interactions of metallodrugs with macromolecular blood components have been recognized as crucial for their biodistribution and efficacy [17-21]. In particular, ruthenium complexes of known anticancer activity have been shown to interact with human serum albumin (HSA) [20, 22, 23], and with apotransferrin (ATf) [24, 25]. We have shown that such protein-drug interactions also take place with ruthenium-chloroquine complexes with antimalarial activity [26]. In this paper we report the results of a study on the binding of compounds 1-6 to HSA and to ATf through monitoring their ability to quench the inherent fluorescence of the tryptophan residues naturally present in the two proteins studied.
Results and Discussion
We recently reported the synthesis and characterization of complexes 1-6, together with activity data against L. major and T. cruzi [16]. All the compounds in this series display in vitro activity against the two parasites and low toxicity toward mammalian cells. In general, the Ru-KTZ combination is more active and/or selective than the individual components of the complexes. The most active derivative against both parasites is complex 3 and this was ascribed, in part, to the presence of the chloride ligands, which readily exchange with water to yield the active species [RuII(η6-p-cymene)(H2O)2(KTZ)]2+ (3a). This cationic compound contains labile water molecules, which may in turn be exchanged by donor atoms of relevant biomolecules present in the medium, e.g. DNA or proteins. The mechanism of biological action of compound 3 is thought to be analogous to the one previously proposed by us for the related complex RuCl2(CTZ)2, also highly active against T. cruzi [13]. The therapeutic pathway involves transit of the active cationic derivative [Ru(H2O)2(CTZ)2]2+ into the parasite cell, where CTZ dissociates from the metal, allowing a dual target action: dissociated CTZ exerts its known SBI action, while the Ru-containing residue binds covalently to the DNA of the parasite. The combined effects account for the high activity and low toxicity of RuCl2(CTZ)2 and by analogy, of complexes 3/3a [14].
It is also of interest to investigate possible mechanisms of transport of the new Ru-based drugs to their biological targets. It is known that the distribution, excretion, and efficacy of a drug are related to its ability to interact with serum proteins [17-21]. Human serum albumin (HSA) is the most abundant blood plasma protein, and it is capable of reversibly binding metallodrugs.21 Transferrin, in turn, is the protein primarily responsible for transporting iron throughout the body, and it can also bind metallodrugs reversibly [23-25]. In particular, ruthenium complexes with anticancer [20, 22, 23] or antimalarial [26] properties have been shown to interact with human serum albumin (HSA) and with apotransferrin (ATf). We have now investigated the interactions of HSA and ATf with complexes 1-6 by use of a fluorimetric titration technique.
Fluorimetric titrations provide information on the interaction of small molecules with proteins; quenching of the tryptophan fluorescence, in particular, has been frequently used to study proteins and protein complexes with drugs. Two different quenching mechanisms may account for the decrease in fluorescence intensity of a protein [27-29]. Dynamic (or collisional) quenching is observed when a compound collides with the excited-state fluorophore absorbing the energy and bringing the fluorophore back to the ground state. Static quenching, on the other hand, occurs when a molecule binds to a protein near the fluorophore, thereby producing a dark complex that does not emit light. Determining which of the two mechanisms is the cause of the quenching provides evidence for the kind of interaction taking place between the drug and the protein. Static quenching, in particular, provides evidence that binding interactions are taking place between the drug and the protein at a site near the tryptophan residue, and such binding is suggestive that the protein may be a suitable transporter for the drug.
Interaction of Compounds 1-6 with HSA
Upon excitation of the tryptophan residue at 295 nm, fluorimetric titrations were monitored at 338 nm to saturation using complexes 1-6. As an example, Figure 1(a) shows the spectra resulting from the interaction of HSA with complex 3. Increasing concentrations of complex 3 notably reduce the intrinsic fluorescence emission of HSA and then quench it, without shifting the maximum or changing the shape of the peaks. All other Ru-KTZ complexes studied quenched the HSA fluorescence in a similar manner after addition of about twenty equivalents, yielding spectra analogous to those in Fig. 1(a) (data not shown). The normalized HSA fluorescence intensity titration curves for complexes 1-6, monitored at 338 nm, are collected in Fig. 1(b).
In order to gain insight into the quenching mechanism, the fluorescence quenching data for complexes 1-6 were analyzed by using the Stern-Volmer equation [30].

where F0 and F are the fluorescence intensities of HSA in the absence and in the presence of the quencher, [Q] is the concentration of the quencher and KSV is the Stern-Volmer quenching constant, which may be interpreted differently depending on the type of quenching envisaged [29]. For dynamic quenching, KSV = kqτ0, where kq is the bimolecular quenching constant and τ0 is the lifetime of the fluorophore [28]. A value of the bimolecular quenching constant (kq) of 1 × 1010 M-1s -1 may be considered as the upper limit for diffusion-controlled collisional quenching [31]. Values of kq greater than this limit may be taken as evidence of static quenching, and therefore of drug-protein adduct formation.
Figure 2(a) shows the Stern-Volmer plots for the fluorescence quenching of HSA caused by complexes 1-6. Note that the plots for all complexes except for 1 display a positive deviation (upward curvature), which may be taken as evidence for static quenching. The values of the corresponding Ksv constants were determined from the initial slopes of the Stern-Volmer plots and are collected in Table 1. Values of kq were also calculated for complexes 1-6, assuming only dynamic quenching and using a lifetime (τo) of 6.0 ns for the tryptophan residue of HSA [32]; they are also included in Table 1. We note that all the Ru-KTZ complexes display values of kq higher than the accepted limit for diffusion controlled bimolecular quenching constant, providing further evidence for static quenching taking place. The combined data are thus strongly indicative that the Ru-KTZ complexes 1-6 bind to HSA to form dark protein-complex adducts.

The fluorescence quenching data obtained from the interaction of complexes 1-6 with HSA were further analyzed in order to obtain binding parameters, by using the equation
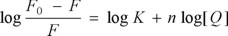
where F0 and F are the fluorescence intensity in the absence and in the presence of the Ru-KTZ complexes, respectively.33 Plots of log [(F0 -F)/F] vs. log [Q] gave straight lines whose slope provided n, the number of binding sites, while the intercept allowed the calculation of the values for the apparent binding constant K. The plot for complex 3 is shown in Fig. 2(b) as an example; data for all complexes are collected in Table 1. The values obtained for K indicate a fairly high affinity of the Ru-KTZ complexes for HSA, in the same order of magnitude as those observed by us for related Ru-chloroquine complexes with antimalarial activity [26].
Interaction of Compounds 1-6 with ATf
Fluorimetric titration of human apotransferrin with complexes 1-6 demonstrated a similar interaction to that with HSA. As an example, Figure 3(a) shows the titration spectra of ATf with increasing amounts of complex 3. Tryptophan fluorescence emission was monitored at 330 nm for complexes 1-6 (Figure 3(b)) and saturation quenching was observed after addition of about 25 equivalents of the complexes. The corresponding Stern-Volmer plots are shown in Figure 4(a), where the upper curvature typical of static quenching is again observed.
The values of Ksv collected in Table 2 were obtained from the initial linear section of the Stern-Volmer plots. Using the ATf tryptophan fluorescent lifetime of 3.34 ns [34], kq values were calculated and they are included in Table 2. As in the case of HSA, the values of the bimolecular quenching constants are higher than the limit for dynamic quenching and are indicative of the formation of a protein-drug adduct. The apparent binding constants and the number of binding sites were also calculated for each complex, using the same equation as for HSA, and are also contained in Table 2; the plot for complex 3 is shown in Fig. 4(b) as an example. As in the case of HSA the K values suggest a fairly strong affinity of complexes 1-6 for ATf, comparable to that observed for related bioactive RuII complexes [26].

Consideration of inner-filter and solvent effects
Absorbance spectral measurements showed that complexes 1-6 all absorb at 295 nm, the excitation wavelength of tryptophan. The attenuation of the excitation beam produced by such absorption due to the Ru-KTZ complexes results in a fluorescence intensity decrease known as the inner-filter effect [29]. A correction was therefore applied to all our fluorescence data, by use of Parker's equation, which has proven to be adequate for this purpose [35].

where Fcorr and Fobs are the corrected and observed fluorescence intensities, ε is the molar extinction coefficient at the excitation wavelength, c is the concentration of the Ru-KTZ complex and d1 and d2 are cuvette dimensions. Since the concentrations of the complexes were low, most compounds had maximum correction factors in the range of 1.01-1.04. Although overall inner-filter effects were minimal, corrected values were used in all our data treatment and analysis.
Also, since DMSO solutions of complexes 1-6 were used in the fluorimetric measurements, control titration experiments for both HSA and ATf were performed using neat DMSO as the possible quencher. No appreciable change in the fluorescence intensity of either protein was observed due to quenching by this solvent.
Conclusions
The interactions of complexes 1-6 with human serum albumin and apotransferrin were evaluated as possible mechanisms of drug transport. Fluorimetric titrations of complexes 1-6 with HSA were analyzed by use of Stern-Volmer plots that yielded Ksv and kqvalues in the order of 104 M-1 and 1013 M-1s-1 respectively. These calculated kq values are 3-4 orders of magnitude higher than the limit of diffusion controlled bimolecular quenching constant expected for collisional quenching. Static quenching must then be operating in this case, indicating the formation of Ru complex-protein adducts. Similar results were observed for the interaction of complexes 1-6 with human apotransferrin. The combined results indicate that both HSA and ATf are possible effective transporters for the new Ru-KTZ antiparasitic drugs.
Experimental
Materials
Human serum albumin (HSA) and human apotransferrin (ATf) were obtained from Sigma. Complexes 1-6 were synthesized as described previously [16]. Solvents were purified by use of a PureSolv purification unit from Innovative Technology; all other chemicals were used as received. Solutions of HSA and ATf were prepared in 10 mM phosphate buffer at pH 7.4. A low concentration of phosphate buffer was used in order to minimize quenching by the phosphate ion [36]. HSA solutions were prepared using a molecular weight of 66.437 kDa. The concentration of the ATf solution was determined from the absorbance at 278 nm using 93,000 M-1cm -1 as the molar extinction coefficient (ε) [37]. Solutions of complexes 1-6 (8 mM) were prepared in DMSO.
Apparatus
Steady-state fluorescence measurements were performed on a PTI's QuantaMaster 40 UV-VIS Spectrofluorometer equipped with a high efficiency continuous Xenon arc lamp and a slit width of 2 nm. Absorption spectra were recorded on an Agilent 8453 diode-array spectrophotometer. One cm quartz cells were used for measurements.
Procedures
Fluorimetric Titrations
Fluorescence spectra were recorded from 300 nm to 450 nm upon tryptophan excitation at 295 nm. Fluorimetric titrations were performed by titrating a 2 mL sample of either 10 µM HSA or 3.8 µM ATf with incremental 2.5 µL additions of 8 mM titrant until saturation was reached. Fluorescence intensities were monitored at 338 nm for HSA and at 330 nm for ATf. All titrations were performed at room temperature (27 ºC) and averages between two trials are reported.
Controls
In order to correct for possible fluorescence emitted by the compounds, background scans for compounds 1-6 were obtained by mixing a solution of each complex in DMSO (50 µL, 8 mM) with phosphate buffer (2 mL, 10 mM, pH 7.4). This measured fluorescence was subtracted from all fluorescence intensities during the titrations. Samples of HSA (2.0 mL, 10 µM) and ATf (2.0 mL, 3.8 µM) in phosphate buffer (10mM) were titrated by repeated additions (2.5 µL) of neat DMSO to test for possible quenching effect from the solvent. Inner filter effects coming from the absorption of the compounds were corrected by using Parker's equation [35]. The molar absorptivities of complexes 1-6 were calculated from the corresponding absorbances at 295 nm at a concentration of 8mM in 2.0 mL of 10mM phosphate buffer.
Acknowledgement
We thank the NIH for financial support through Grant # 5SC1GM089558-03.
References
1. WHO Control of leishmaniases. Tech. Rep. Ser. Vol. 949, 2010. [ Links ]
2. WHO Report of the consultative meeting on Cutaneous Leishmaniasis, 2007. [ Links ]
3. den Boer, M.; Argaw, D.; Jannin, J.; Alvar, J. Clin Microbiol Infect 2011, 17, 1471. [ Links ]
4. WHO Control of Chagas disease. Tech. Rep. Ser. Vol. 905, 2002. [ Links ]
5. Kirchhoff, L. V. Adv Parasitol. 2011, 75, 1. [ Links ]
6. Croft, S. L.; Barrett, M. P.; Urbina, J. A. Trends Parasitol. 2005, 21, 508. [ Links ]
7. Drug Resistance in Leishmania Parasites: Consequences, Molecular Mechanisms and Possible Treatments; Ponte-Sucre, A.; Diaz, E.; Padron-Nieves, M., Eds.; Springer: Vienna, 2012. [ Links ]
8. Urbina, J. A. Drug Fut. 2010, 5, 409. [ Links ]
9. Espuelas, S.; Plano, D.; Nguewa, P.; Font, M.; Palop, J. A.; Irache, J. M.; Sanmartin, C. Curr. Med. Chem. 2012, 19, 4259. [ Links ]
10. Sánchez-Delgado, R. A.; Anzellotti, A. Mini-Rev. Med. Chem. 2004, 4, 23. [ Links ]
11. Sánchez-Delgado, R. A.; Anzellotti, A.; Suárez, L. In Met Ions Biol Syst; Siegel, H., Siegel, A., Eds.; Marcel Dekker, Inc.: New York, 2004; Vol. 41, p 379. [ Links ]
12. Urbina, J. A. Mem. Inst. Oswaldo Cruz 2009, 104, 311. [ Links ]
13. Sánchez-Delgado, R. A.; Lazardi, K.; Rincon, L.; Urbina, J. A.; Hubert, A. J.; Noels, A. N. J. Med. Chem. 1993, 36, 2041. [ Links ]
14. Sánchez-Delgado, R. A.; Navarro, M.; Lazardi, K.; Atencio, R.; Capparelli, M.; Vargas, F.; Urbina, J. A.; Bouillez, A.; Noels, A. F.; Masi, D. Inorg. Chim. Acta 1998, 275-276, 528. [ Links ]
15. Martínez, A.; Carreon, T.; Iniguez, E.; Anzellotti, A.; Sánchez, A.; Tyan, M.; Sattler, A.; Herrera, L.; Maldonado, R. A.; Sánchez-Delgado, R. A. J. Med. Chem. 2012, 55, 3867. [ Links ]
16. Iniguez, E.; Sánchez, A.; Carreon, T.; Martínez, A.; Vasquez, M. A.; Sattler, A.; Sánchez-Delgado, R. A.; Maldonado, R. A. J Biol Inorg. Chem. 2013, doi: 10.1007/s00775-013-1024-2. [ Links ]
17. Ascone, I.; Messori, L.; Casini, A.; Gabbiani, C.; Balerna, A.; Dell'Unto, F.; Congiu, C. A. Inorg. Chem. 2008, 47, 8629. [ Links ]
18. Groessl, M.; Terenghi, M.; Casini, A.; Elviri, L.; Lobinski, R.; Dyson, P. J. J. Anal. At. Spectrom. 2010, 25, 305. [ Links ]
19. Shmykov, A. Y.; Filippov, V. N.; Foteeva, L. S.; Keppler, B. K.; Timerbaev, A. R. Anal. Biochem. 2008, 379, 216. [ Links ]
20. Espósito, B. P.; Najjar, R. Coord. Chem. Rev. 2002, 232, 137. [ Links ]
21. Timerbaev, A. R.; Hartinger, C. G.; Aleksenko, S. S.; Keppler, B. K. Chem. Rev. 2006, 106, 2224. [ Links ]
22. Messori, L.; Orioli, P.; Vullo, D.; Alessio, E.; Iengo, E. Eur. J. Biochem. 2000, 267, 1206. [ Links ]
23. Stepanenko, I. N.; Casini, A.; Edafe, F.; Novak, M. S.; Arion, V. B.; Dyson, P. J.; Jakupec, M. A.; Keppler, B. K. Inorg. Chem. 2011, 50, 12669. [ Links ]
24. Frasca, D. R.; Gehrig, L. E.; Clarke, M. J. J. Inorg. Biochem. 2001, 83, 139. [ Links ]
25. Kratz, F.; Hartmann, M.; Keppler, B.; Messori, L. J. Biol. Chem. 1994, 269, 2581. [ Links ]
26. Martinez, A.; Suarez, J.; Shand, T.; Magliozzo, R. S.; Sánchez-Delgado, R. A. J. Inorg. Biochem. 2011, 105, 39. [ Links ]
27. Divsalar, A.; Bagheri, M.; Saboury, A.; Mansoori-Torshizi, H.; Amani, M. J. Phys. Chem. B 2009, 113, 14035. [ Links ]
28. Kandagal, P.; Ashoka, S.; Seetharamappa, J.; Shaikh, S.; Jadegoud, Y.; Ijare, O. J. Pharm. Biomed. Anal. 2006, 41, 393. [ Links ]
29. Samworth, C.; Esposti, M.; Lenaz, G. Eur. J. Biochem. 1988, 171, 81. [ Links ]
30. Stern, O.; Volmer, M. Phys. Z. 1919, 20, 103. [ Links ]
31. Lakowicz, J. In Principles of Fluorescence Spectroscopy; Springer: U.S., 2006; Vol. 3rd Edition, p 282. [ Links ]
32. Lakowicz, J.; Weber, G. J. Biophys. 1973, 12, 591. [ Links ]
33. Froehlich, E.; Mandeville, J. S.; Jennings, C. J.; Sedaghat-Herati, R.; Tajmir-Riahi, H. A. J. Phys. Chem. B 2009, 113, 6986. [ Links ]
34. James, N.; Ross, J.; Mason, A.; Jameson, D. Prot. Sci. 2010, 19, 99. [ Links ]
35. Schirmer, R. In Modern Methods of Pharmaceutical Analysis; CRC Press: 1990; Vol. 2nd, p 213. [ Links ]
36. Maruthamuthu, M.; Selvakumar, G. Proc. Indian. Acad. Sci. Chem. Sci. 1995, 79. [ Links ]
37. Du, H.; Xiang, J.; Zhang, Y.; Tang, Y.; Xu, G. J. Inorg. Biochem. 2008, 102, 146. [ Links ]

