










Services on Demand
Journal
Article
 English (pdf)
English (pdf)
 Article in xml format
Article in xml format Article references
Article references
 Send this article by e-mail
Send this article by e-mailIndicators
-
 Cited by SciELO
Cited by SciELO -
 Access statistics
Access statistics
Related links
-
 Similars in
SciELO
Similars in
SciELO
Share

 Permalink
PermalinkJournal of the Mexican Chemical Society
Print version ISSN 1870-249X
J. Mex. Chem. Soc vol.57 n.3 Ciudad de México Jul./Sep. 2013
Article
Transfer Hydrogenation Reactions of Photoactivatable N,N'-Chelated Ruthenium(II) Arene Complexes
Soledad Betanzos-Lara,1,2 Abraha Habtemariam,*,1 and Peter J. Sadler1
1 Department of Chemistry, University of Warwick, Coventry, UK CV4 7AL.
2 Current address: Massachusetts Institute of Technology, 77 Massachusetts Avenue, Cambridge, MA 02139, U.S.A. a.habtemariam@warwick.ac.uk; P.J.Sadler@warwick.ac.uk
Received March 27, 2013
Accepted June 18, 2013
Abstract
We show that the reaction of RuII arene chlorido complexes of the type [(η6-arene)Ru(N,N')Cl]+ arene = p-cymene (p-cym), hexamethylbenzene (hmb), indane (ind), N,N' = bipyrimidine (bpm) and 1,10-phenanthroline (phen) with excess sodium formate generates a very stable formate adduct through spontaneous hydrolysis of the Ru-Cl bond at 310 K and pH* = 7.0. The formate adducts are also produced when RuII arene pyridine complexes of the type [(η6-arene)Ru(N,N')(Py)]2+ (where Py = pyridine), are irradiated with UVA (λirr = 300-400 nm) or visible light (λirr = 400-660 nm) under the same conditions. The RuII arene formato adducts do not catalyse the reduction of acetone through transfer hydrogenation. However, all the complexes (except complex 2 which contains phen as the chelating ligand) can catalyse the regioselective reduction of NAD+ in the presence of formate (25 mol equiv) in aqueous solution to form 1,4-NADH. The catalytic activity is dependent on the nature of the chelating ligand. Most interestingly, the regioselective reduction of NAD+ to 1,4-NADH can be also specifically triggered by photoactivating a RuII arene Py complex.
Key Words: Ruthenium, arene, pyridine, hydride, NAD+/NADH, photoactivation.
Resumen
En el presente trabajo demostramos que la interacción de complejos areno cloruro de Ru(II) del tipo [(η6-areno)Ru(N,N')Cl]+ donde areno = p-cimeno (p-cym), hexametilbenceno (HMB), indano (ind); N,N' = bipirimidina (bpm) y 1,10-fenantrolina (phen) con un exceso de formato de sodio genera un aducto formato muy estable a través de la hidrólisis espontánea del enlace Ru-Cl a 310 K y pH = 7.0*. Los aductos de formato también se producen cuando un complejo areno piridina de Ru(II) del tipo [(η6-areno)Ru(N,N')(Py)]2+ (donde Py = piridina), se irradia con UVA (λirr = 300-400 nm) o luz visible (λirr =400-660 nm) bajo las mismas condiciones. Los aductos areno formato de Ru(II) no catalizan la reducción de acetona a través de transferencia de hidrógeno. Sin embargo, todos los complejos (excepto el complejo 2 que contiene phen como el ligante quelante) pueden catalizar la reducción regioselectiva de NAD+ en presencia de formato (25 equiv) en solución acuosa para formar 1,4-NADH. La actividad catalítica depende de la naturaleza del ligante quelante. De manera notable, la reducción regioselectiva de NAD+ a 1,4-NADH también puede ser iniciada específicamente por medio de la fotoactivación de un complejo de areno piridina de Ru(II).
Palabras clave: Rutenio, Areno, Piridina, Hidruro, NAD+/NADH, Fotoactivación.
Introduction
The observation that some RuII arene complexes can form stable hydride complexes [1-3] in aqueous solution using formate as a hydride source [4-6], has opened up a new avenue for investigation of water-soluble organometallic complexes as catalysts for transfer hydrogenation. The area has attracted increasing interest related to environmentally sustainable processing, simple product separation, and pH dependent selectivity in aqueous media [7, 8]. For example, the RuII complexes [(η6-C6Me6)Ru(bpy)(OH2)]2+ and [(η6-C6Me6)Ru(phen)Cl]+ where bpy is 2,2'-bipyridine and phen is 1,10-phenanthroline and other related complexes [9] have been shown to catalyse the reduction of ketones (such as cyclohexanone and acetophenone) to alcohols and imines [10-12]; although the conditions for optimum turnover are (usually) not biologically compatible [5, 13]. It has been noted that the catalytic activity usually requires the presence of a labile coordination site on the RuII centre and/or arene displacement [14, 15] and that the nature of all the coordinated ligands can have a pronounced effect on the catalytic properties of these complexes. This observation has led to the development of a wide range of synthetic routes to complexes as catalytic precursors containing various substituted arenes together with other ligands such as halides, carboxylates, amines, oxygen or nitrogen chelating groups, Schiff bases, carbenes, phosphines, alkyl, and aryl groups [16-18].
In the field of biocatalysis, RhIII pentamethylcyclopentadienyl [19, 20] and RuII arene complexes [6] have been shown to catalyse the reduction of β-nicotinamide adenine dinucleotide (NAD+) in the presence of formate as the hydride source. This reduction is regioselective, giving the biologically relevant 1,4-NADH isomer and in the case of the RhIII derivative, it was further shown that it can drive enzymatic reactions relying on NADH as a cofactor [21]. In the present work, hydride-transfer reactions of a series of RuII arene halido complexes that region-selectively reduce NAD+ in the presence of formate under biologically relevant conditions are described. It is also shown that this reaction can be specifically photo-triggered when a pyridine complex is irradiated with UVA (λirr = 300-400 nm) or visible light (λirr = 400-660 nm).
Results
Reactions of RuII Arene Chlorido Complexes with Sodium Formate
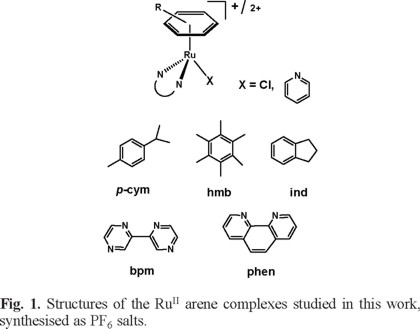
The RuII arene chlorido complexes 1-4 of the form [(η6-arene)Ru(N,N')Cl]+ where arene = p-cymene (p-cym), hexamethyl benzene (hmb), indane (ind); and N,N' = 2,2'-bipyrimidine (bpm), and 1,10-phenathroline (phen) used in this study were previously reported by us [22]. The complexes are listed in Table 1 and their general structures are shown in Figure 1. The potential of these complexes as transfer hydrogenation catalysts in the presence of sodium formate (as a source of hydride) was investigated. All the reactions were carried out in NMR tubes using 3.0 mM solutions of the complexes (in 90% H2O/10% D2O) and changes in the spectra were followed over 24 h at 310 K. The initial pH* (pH meter reading in D2O solutions) of the reaction mixture in the presence of formate (25 mol equiv) was 6.9-7.2. The 1H NMR spectra of complexes 1-4 initially contained one major set of peaks corresponding to the chlorido species followed by a second set of peaks that appeared and increased in intensity with time. The new set of peaks has the same chemical shifts as those of the aqua adducts under the same conditions; the aqua adducts were independently prepared by treatment of the chlorido complexes with AgNO3 in water at ambient temperature overnight and removal of the precipitate (AgCl) by filtration. A third set of peaks in the 1H NMR spectra was also observed over time and these were attributed to the formation of a RuII formato adduct, [(η6-arene)Ru(N,N')O2CH]+, in each case. Peaks assigned to the bound formate in all compounds are high-field shifted in comparison to those of free formate (8.40 ppm under the same conditions; 310 K and pH* = 7.0). The 1H NMR peak for the formate adduct of complex 1 has a chemical shift of 7.29 ppm whereas for complexes 2, 3, and 4, the corresponding singlet is observed at 7.65, 7.68, and 7.68 ppm, respectively. The mass-to-charge ratios and isotopic models obtained from HR-MS spectra are consistent with the formation of the formato complexes, however no evidence for the formation of the Ru-H species in the high-field region of the 1H NMR spectrum was detected, Table 2. In most cases, the reactions reached equilibrium within the first hour after mixing. No further changes in the amount of species present were observed after 24 h, as quantified by integration of the peaks in the 1H NMR spectra. No significant changes in the initial pH* of the mixture were observed at the end of the reaction. Figure 2 shows the progress of the reaction of [(η6-hmb)Ru(bpm)Cl]+ (3) followed by 1H NMR spectroscopy as a generic example.



When the N,N'-chelating ligand was varied (bpm in complex 1 and phen in 2) the time needed for formato-complex formation to reach equilibrium was longer for 1 than for 2, Table 3. Varying the arene and keeping the chelating ligand as bpm resulted in a decreasing reaction rate, ind (4) > p-cym (1) > hmb (3). The reaction of complex 2 was found to give the lowest yield. To ascertain whether the RuII arene formato complexes may be directly involved in transfer hydrogenation reactions, the reduction of acetone to form iso-propanol was investigated. The addition of 10 mol equiv of acetone to the equilibrated reaction mixture resulted in no product, suggesting that the formato adduct is inert under these conditions.

In order to study the possibility of generating hydride species (Ru-H) in solution, the formato complex [(η6-pcym)Ru(bpm)(O2CH)][PF6] (5), Figure 3, was synthesised as a PF6 salt in good yield (64%). It was fully characterised by 1D and 2D 1H NMR methods as well as HR-MS. Compared to its chlorido analogue (complex 1), the 1H NMR resonances of complex 5 are high-field-shifted by ca. 0.3 ppm. The binding of the formate ligand to the RuII centre was confirmed by the appearance of a sharp singlet at 7.66 ppm (compared to free formate at 8.40 ppm under the same conditions, 310 K and pH = 7.2), Figure S1. With the purpose of investigating the hydrolysis behaviour, the changes in the 1H NMR spectrum of a freshly-made 100 μM solution of complex 5 (90% H2O/10% D2O) were followed for 24 h at 310 K. The 1H NMR spectrum of complex 5 initially contained one major set of peaks (assignable to formato species) followed by a second set of peaks which increased in intensity over time. The new set of peaks had the same chemical shifts as those of the aqua adduct under the same conditions (100 μM solution in 90% H2O/10% D2O at 310 K and pH = 7.2). Figure S2 shows the changes of the 1H NMR spectrum during the hydrolysis reaction of complex 5. The mass-to-charge ratio and isotopic model obtained from HR-MS spectra were consistent with the formation of the aqua adduct. The extent of aquation for complex 5 reached 68% after 24 h. No arene loss (p-cym) and no formation of Ru-H species were detected over this period.

The kinetics of the aquation was also studied by UV-Vis spectroscopy. The dissolution of [(η6-p-cym)Ru(bpm)(O2CH)][PF6] (5) in H2O at 310 K gave rise to ligand substitution reactions as indicated by the concomitant changes in the UV-Vis absorption bands. The time evolution spectrum for complex 5 is shown in Figure S3. The time dependence of the absorbance for complex 5 at λ = 288 nm followed pseudo first-order kinetics, Figure 4.The corresponding rate constant (k × 10-3, min-1) and half-life (t1/2, min) of 15.3 ± 0.58 and 45.3, respectively, were determined (the errors quoted are fitting errors).

Regioselective Reduction of NAD+ in the Presence of Formate
The RuII arene complexes 1-4 were studied as catalyst for the regioselective reduction of β-nicotinamide adenine dinucleotide (NAD+) to form β-nicotinamide adenine dinucleotide (1,4-NADH) in the presence of sodium formate (as a source of hydride) by means of multidimensional 1H NMR spectroscopy over 24-48 h at 310 K. The structures and numbering scheme for NAD+, 1,4-NADH and 1,6-NADH are shown in Figure S4. The pH* of the RuII arene chlorido solutions prior to the addition of NAD+ was 6.9-7.2. A decrease of the pH* value to 5.2-5.8 was recorded after addition of NAD+. Figure 5 shows the 1H NMR spectra of [(η6-hmb)Ru(bpm)Cl]+ (3) in 90% H2O/10% D2O (3.0 mM) at 310 K in the presence of an excess of sodium formate and NAD+ (molar ratios 1:25:1, respectively) after 300 min of reaction as a generic example. The spectrum initially contained three major sets of peaks corresponding to the chlorido complex [(η6-hmb)Ru(bpm)Cl] (3), the aqua [(η6-hmb)Ru(bpm)(OH2)]2+, and the formato adduct [(η6-hmb)Ru(bpm)(O2CH)]+ at equilibrium. Upon addition of NAD+ (1 mol equiv), some changes in the 1H NMR spectrum were observed within the first 30 min of reaction. The changes suggest the fast regioselective reduction of NAD+ to 1,4-NADH as indicated by a decrease in the intensity of the signals of free NAD+ (H2 at 9.38 ppm) and the new peaks assignable to 1,4-NADH (H2 at 6.92 ppm and H4a/H4b at 2.70 ppm). The 1H NMR spectra within the first 30 min of reaction also reveal the emergence of a second singlet around 7.20 ppm that could be tentatively assigned to the H2 of 1,6-NADH (as a side product). It was noticed that after a total of ca. 300 min, all of NAD+ had been fully consumed. The progress of the reaction was further monitored for a further 24 h after the initial amount of NAD+ had been consumed. The sharp singlet which appeared at around -7.49 ppm (which can be assigned to a Ru-H) [30] Figure 6, increased in intensity over time.


Since the accumulation of 1,4-NADH seemed to be linked with the formation of Ru-H species [30], an extra mol equiv of NAD+ was added to the reaction mixture at this stage (t = 560 min). The 1H NMR spectrum after ca. 5 min showed the disappearance of the Ru-H peaks, Figure S5. As the reaction progressed, an increase in the intensity of the 1,4-NADH signals was observed with the concomitant decrease in the intensity of the signals for NAD+. Also the signals of the Ru-H species were restored. A decrease in the intensity of the peaks for the formate adduct [(η6-hmb)Ru(bpm)(O2CH)]+ following the addition of extra amounts of NAD+ was also detected. Around t = 504 min (after the addition of extra NAD+), the second mol equiv of NAD+ was again fully consumed. The Ru-H signal at -7.49 ppm reached maximum intensity. Complex [(η6-ind)Ru(bpm)Cl]+ (4) was also found to catalyse the reduction of NAD+ to 1,4-NADH (as observed by 1H NMR spectroscopy) in a similar fashion to complex 3 under the same conditions (310 K and pH* = 5.2). Complex [(η6-p-cym)Ru(phen)Cl]+ (2) did not catalyse the hydride-transfer reaction under these conditions.
Reactions of RuII Arene Pyridine Complexes with Sodium Formate upon Visible Light Photoirradiation
The possibility that the photoactivatable RuII arene pyridine complexes studied by us previously [23] could be involved in transfer hydrogenation reactions in the presence of formate was also investigated. The [(η6-arene)Ru(N,N')(Py)]2+ complexes are listed in Table 4 and their general structures are shown in Figure 1. All the reactions were carried out in NMR tubes as 3.0 mM (90% H2O/10% D2O) solutions and followed by 1H NMR spectroscopy at different stages of photoirradiation with visible light (λirr = 400-660 nm) for 12 h at 310 K. The initial pH* of solutions of the RuII arene pyridine complexes in the presence of sodium formate (25 mol equiv) was in the range of 7.0-7.4. Upon photoirradation with visible light at 310 K, a new set of peaks began to appear and were assigned to an aqua adduct along with the peaks for the released Py ligand. Soon afterwards, a second new set of peaks corresponding to the formation of the formato adduct began to appear. The new set of peaks increased in intensity with time. The pH* of the irradiated solutions at the end of the photoirradiation experiment was ca. 7.0 in all cases. Typical spectra recorded during the course of the photoirradiation are shown for [(η6-p-cym)Ru(bpm)(Py)]2+ (7) in Figure S6. Peaks assigned to the formato adducts have identical chemical shifts to those formed directly from the chlorido complexes 1-4 (no irradiation). The mass-to-charge ratios and isotopic models obtained from HR-MS spectra of the resulting irradiated samples of the Py complexes 6-10 are also identical to those of the formato adducts of complexes 1-4 . The HR-MS spectra of the irradiated solution of complex [(η6-ind)Ru(bpy)(Py)]2+ (10) (for which no chlorido analogue was investigated) gave observed peaks expected for [(η6-ind)Ru(bpy)(O2CH)]+. For complexes 6, 7 and 10 less than ca. 50% of the original RuII arene pyridine complex had been photoconverted to the corresponding aqua adduct after ca. 10 h of continuous visible light photoirradiation. A change of arene (i.e. from p-cym to hmb) modified the extent of the photoreaction as well as the time needed to achieve the photoconversion. The fastest reactions were those for complexes [(η6-hmb)Ru(bpm)(Py)]2+ (9) and [(η6-ind)Ru(bpm)(Py)]2+ (10).6 The aqua adducts generated upon photoirradiation where found to react simultaneously with the excess formate present in the solution to generate the corresponding formato complexes. Table 5 lists the percentage of species detected by 1H NMR after 12 h of continuous visible light photoirradiation (λirr = 400-660 nm) of complexes 6-10. The ability of these complexes to effect transfer hydrogenation under photoactivation conditions was assessed by the addition of 10 mol equiv. of acetone to the reaction mixture as has been described earlier (vide supra). However, no reduction of the acetone to iso-propanol was observed.


Regioselective Reduction of NAD+ by RuII Arene Complexes in the Presence of Formate upon UVA Photoirradiation
Complex [(η6-hmb)Ru(bpm)(Py)]2+ (8), was selected to explore the possibility of photo-triggering the regioselective reduction of NAD+ to 1,4-NADH in the presence of sodium formate. The interactions were studied by means of multidimensional 1H NMR spectroscopy. All the reactions were carried out in NMR tubes in a 90% H2O/10% D2O solution and followed at different stages of photoirradiation with UVA (λirr = 300-400 nm) at 310 K. The initial pH* of the solution (prior to the addition of NAD+) was 7.6; once NAD+ was, a decrease to pH 5.8 was registered. Figure 7 shows the progress of the reaction for complex 8 within the first 330 min of photoirradiation. The phenomena of Py release and formation of aqua adduct previously described by us [23] was observed. An additional set of peaks increased in intensity just after the first indication of aqua adduct being formed was detected; this new set of peaks corresponds to the formato adduct [(η6-hmb)Ru(bpm)(O2CH)]+ generated as described above (vide supra). During the first 130 min of photoirradiation with UVA (λirr = 300-400 nm) some additional changes in the 1H NMR spectrum were noticed simultaneously. These changes resemble those observed for the reaction of the chlorido complex [(η6-hmb)Ru(bpm)Cl]+ (3) with NAD+ after ca. 300 min. The changes point again to the relatively fast reduction of NAD+ to 1,4-NADH, as indicated by a decrease in the intensity of the signals corresponding to free NAD+ and the new peaks assignable to 1,4-NADH. The overall changes in the 1H NMR spectra indicate that although the initial amount of NAD+ is not fully consumed in this case (as opposed to the chlorido analogue), the reaction to generate 1,4-NADH is relatively faster. Furthermore, when a control solution of the pyridine RuII arene complex 8, NAD+ and formate under the same conditions was kept in the dark (as a control), no reaction was observed. After ca. 300 min, the photoirradiation was suspended and the mixture was allowed to further react in the dark at 310 K. The resulting spectrum is shown in Figure S7. Within the next 300 min (and overall reaction time of ca. 660 min) the appearance of multiple low-intensity signals in the aromatic region was detected. The 1H NMR spectra suggest again 1,6-NADH formation as a side-product (as was also observed for the chlorido complexes 3 and 4) but no evidence of Ru-H species was detected over this period.

Discussion
The interaction of RuII arene chlorido complexes (1-4) with an excess of sodium formate in aqueous solution at 310 K, showed that very stable formato adducts can be formed. This adduct (Ru-O2CH) is generated by the direct substitution of H2O (Ru-OH2) formed in situ via Ru-Cl bond hydrolysis. This labile aqua ligand is displaced by formate which binds through the negatively-charged carboxylate oxygen, as has been previously reported for similar formate and other carboxylate metal complexes [5, 24]. The binding of formate to the RuII centre in these complexes was confirmed by the appearance of a sharp singlet at 7.66 ppm in the 1H NMR spectrum (free formate is at 8.40 ppm). The high field shift has been previously observed for analogous RuII arene formato and acetato complexes [25, 26]. The formato complex [(η6-p-cym)Ru(bpm)(O2CH)]+ (5) had a half-life for hydrolysis of 45 min, and proceeded to a relatively high extent (68.2%). However, no hydrolysis was observed in aqueous solution in the presence of an excess of formate (25 mol equiv). The Ru-O2CH adducts of all of the complexes studied here, did not catalyse the reduction of a ketone to afford an alcohol as exemplified by acetone (10 mol equiv) at 310 K and pH* = 6.9-7.2. This suggests that they are relatively unreactive towards hydride formation and transfer. Previous studies have also demonstrated the formation of stable RuII arene formato complexes in solution and in the solid state [9, 27] as well as non-arene octahedral RuII formato complexes [28]. Surprisingly, the reduction of NAD+ to afford 1,4-NADH was achievable by all the RuII arene bpm complexes in the presence of excess sodium formate (25 mol equiv) in aqueous solution at 310 K. The initial formation of the Ru-H species (suggested in later stages of the reaction to be linked to a critical accumulation of 1,4-NADH, vide infra) is characterised by the appearance of sharp singlets in the high-field region of the 1H NMR spectrum (between -7 and -8 ppm), as it has been observed in similar RuII arene hydride species [19, 29]. A plausible mechanism for the regioselective reduction of NAD+ to 1,4-NADH has been previously suggested for NAD+-models [28, 30]. It should also be noted that hydride-transfer from 1,4-NADH to metal centres is a process that has been shown to occur under similar conditions [30]. Furthermore, if more NAD+ is introduced into the RuII arene catalytic system, the cycle is restarted and then accumulation of 1,4-NADH is again observed (along with the regeneration of the signal for the Ru-H species of complexes 3 and 4 in the negative region of the 1H NMR spectrum), as shown in Figure 8.
The reaction of the half-sandwich RuII arene pyridine complexes (6-10) with an excess of sodium formate (25 mol equiv) in aqueous solution at 310 K and pH* = 6.8-7.1, showed that very stable RuII arene formato adducts (Ru-O2CH) can be formed exclusively upon photoirradiation with UVA (λirr = 300-400 nm) or visible (λirr = 400-660 nm) light. The corresponding adducts (Ru-O2CH) are generated more quantitatively if UVA photoirradiation is used, and they are not susceptible to photodecomposition. The binding of formate to the RuII centre thus generates identical species to those formed from the reaction of the RuII arene chlorido analogues. The corresponding Ru-O2CH adducts formed in solution upon photoirradiation with UVA or visible light, display the same reactivity as those produced by the direct reaction of the analogous chlorido species (vide supra), i.e. they do not catalyse the reduction of the organic substrate acetone (10 mol equiv) under the experimental conditions. The pH* value seems to be a critical factor for hydride-transfer to proceed (being optimal under acidic conditions, vide supra). As observed for the RuII chlorido analogues, the pyridine RuII arene complex [(η6-hmb)Ru(bpm)(Py)]2+ (8) was found to catalyse the conversion of NAD+ into 1,4-NADH in aqueous solution at 310 K in the presence of an excess of sodium formate (molar ratios 1:1:25, respectively) exclusively upon photoirradiation with UVA (λirr = 300-400 nm) or visible light (λirr = 400-660 nm). When a control solution of the RuII arene pyridine complex 8, along with NAD+ and formate was kept in the dark, no reaction was observed. In the proposed mechanism shown in Figure 8 which is supported by published work [11, 18, 28, 30] the first step in the regioselective reduction, involves the photolysis of the corresponding Ru-N(Py) bond in complex 8 (and the selective release of the Py ligand), followed by an almost simultaneous binding of formate to the RuII centre in the Ru-OH2 species formed in situ upon photoirradiation. The rate of the reduction reaction is assumed to be limited by the slow rate of hydride-transfer as proposed for the chlorido complexes 3 and 4. Two main differences between the RuII arene halido complexes (3 and 4) and the RuII arene pyridine complexes are observed. The first is that the aqua adduct of the pyridine complexes is produced exclusively upon UVA or visible light photoirradiation. The second difference is that no Ru-H signal is detected despite the fact that the reduction to 1,4-NADH is indeed observed (by 1H NMR spectroscopy). This could be due not only to the reduced generation of 1,4-NADH (which has been proved to also contribute to the generation of Ru-H species) [30] but also due to possible simultaneous photo-degradation of 1,4-NADH to NAD+ upon photoirradiation by UVA (λirr = 300-400 nm), as it has been recently suggested [31]. It is believed that NAD+, adenosine 5'-diphosphoribose (ADPR) and a second compound, which may be nicotinamide (NA) are the photoproducts resulting from long-time exposures (2 days) of 1,4-NADH to UVA photoirradiation (λirr = 300-400 nm) in water and normal O2 levels form the atmosphere. In that report it was also observed that ADPR and NA emerge exclusively in oxygen-poor conditions.
Concluding Remarks
The interaction of RuII arene chlorido complexes with excess of sodium formate (25 mol equiv) in aqueous solution at 310 K, gave formate adducts upon hydrolysis of the corresponding Ru-Cl bonds. It was observed that the isolated formato complex [(η6p-cym)Ru(bpm)(O2CH)] (5) underwent hydrolysis in aqueous solution at 310 K with a half-life of 45 min and to an extent of more than 65%. The formato adducts can also be generated when a RuII arene pyridine complex is irradiated with UVA (λirr = 300-400 nm) or visible light (λirr = 400-660 nm) under the same conditions (310 K and pH = 7.0). This observation provides further evidence that such RuII arene formate complexes are also stable towards photoirradiation. None of the RuII arene formato adducts catalysed the reduction of acetone, suggesting a decreased reactivity for these complexes at biologically relevant pH values.
Four RuII arene complexes of the type [(η6-arene)Ru(N,N')Cl][PF6] where arene is para-cymene (p-cym, 1), hexamethylbenzene (hmb, 3), indane (ind, 4) and N,N' is 2,2'-bipyrimidine (bmp) were investigated for hydride-transfer reactions. It was found that complexes 3 and 4 can catalyse the regioselective reduction of NAD+ in the presence of formate in water (25 mol excess) to form 1,4-NADH. For these complexes the reaction occurs via the initial formation of a 1H NMR detectable Ru-H (hydride) species where formate is the hydride source. A second reduction product was also detected in the later stages of the reaction as a side-product, 1,6-NADH. The catalytic activity seems to be dependent on the chelating ligand as well as the arene with the hexamethylbenzene (hmb) complex 3, showing the better activity by providing electronic stability during the formation of the Ru-H species and be favoured at lower pH values. It was also discovered that when a critical amount of 1,4-NADH is accumulated in reaction mixture, this later species can act as a hydride source [30].
It was also shown that the regioselective reduction of NAD+ to NADH can be photo-triggered by photo-activating a RuII arene pyridine complex, [(η6-hmb)Ru(bpm)(Py)]2+ (8). In this case, no detectable 1H NMR signals for Ru-H species were observed due to the reduced accumulation of 1,4-NADH (from NAD+) and its decomposition induced by UV light irradiation and it appears in the catalytic cycle only as a non-detectable intermediate.
Experimental
Materials. β-Nicotinamide adenine dinucleotide hydrate (NAD+), sodium formate (NaHCO2), silver nitrate (AgNO3), and potassium hexafluorophosphate (KPF6) were obtained from Sigma-Aldrich. The RuII arene halido complexes [(η6-p-cym)Ru(bpm)Cl][PF6], [(η6-bip)Ru(bpm)Cl][PF6], [(η6-hmb)Ru(bpm)Cl][PF6], [(η6-ind)Ru(bpm)Cl][PF6], and [(η6-p-cym)Ru(phen)Cl)][PF6] where pcym = para-cymene, hmb = hexamethylbenzene, bpm = 2,2'-bipyrimidine and phen = 1,10'-phenanthroline, were synthesised following a method previously described [22, 32]. The RuII arene halido complexes [(η6-(hmb)Ru(en)Cl][PF6], [(η6-ind)Ru(bpy)Cl][PF6], [(η6-ind)Ru(4,4'-Me2-bpy)Cl][PF6], and [(η6-p-ind)Ru(phen)Cl][PF6] were synthesised according to a reported method [33]. The RuII arene pyridine complexes [(η6-p-cym)Ru(bpm)(Py)][PF6]2, [(η6-hmb)Ru(bpm)(Py)][PF6]2, [(η6-ind)Ru(bpm)(Py)][PF6]2, [(η6-p-cym)Ru(phen)(Py)][PF6]2, and [(η6-ind)Ru(bpy)(Py)][PF6]2 were synthesised as previously described [23]. The solvent used for UV-vis absorption spectroscopy was deionised water. The solvents used for 1H NMR spectroscopy were methanol-d4 and D2O from Aldrich unless otherwise stated.
Reactions of RuII Arene Complexes with Sodium Formate (NaHCO2)
The following experiment was carried out under normal ambient light conditions. An excess of NaHCO2 (25 mol equivalent) was added to 3.0 mM solutions of the RuII arene halido complexes in 90% H2O/10% D2O at ambient temperature. The 1H NMR spectra of the resulting solutions were recorded at 310 K at various time intervals over 24-48 h. The pH* of the solutions was recorded at the beginning and at the end of the experiment.
Reactions of RuII Arene Complexes with Sodium Formate (NaHCO2) upon photoirradiation. Aqueous solutions of the RuII arene complexes were photoirradiated at 310 K using the photoreactor LZC 4V Illuminator (Luzchem, Canada) with temperature controller and UVA (λirr = 320-400 nm with a maximum intensity at ~360 nm, 1 J cm-2h-1) or white light lamps (λirr = 400-660 nm providing average light power of 1 J cm-2h-1). These amount to relatively low doses of light (about 15 min in the midday sun).
1H NMR spectra of 3.0 mM (90% H2O/10% D2O) solutions of the RuII arene pyridine complexes in the presence of an excess of NaHCO2 (25 mol equiv) were acquired at different stages of photoirradiation. The pH of the solutions was recorded at the beginning and at the end of the experiment.
Preparation of a RuII Arene Formato Complex. The complex [(η6-p-cym)Ru(bpm)(O2CH)][PF6] was synthesised using a similar procedure previously reported [34]. Using an aluminium-foil-covered flask at room temperature, [(η6-p-cym)Ru(bpm)Cl][PF6] and AgNO3 in a 1:1 mixture of MeOH/H2O (10 mL) were heated under reflux overnight (18 h). Precipitated AgCl was then removed by filtration. Sodium formate (25 mol equiv) was added and the mixture was left stirring for 30 min at ambient temperature. The volume was reduced by rotary evaporation and 2-5 mol equiv of KPF6 was added. The precipitate that formed was collected by filtration and washed with portions of Et2O/MeOH and dried overnight under vacuum, resulting in a microcrystalline product. Details of the amounts of reactants, volumes of solvents mixture, colour changes, and nature of the product are described below.
[(η6-p-cym)Ru(bpm)(O2CH)][PF6] (5). [(η6-p-cym)Ru(bpm)Cl][PF6] (0.10 g, 0.17 mmol), AgNO3 (0.03 g, 0.17 mmol), NaHCO2 (0.10 g, 2.94 mmol) and KPF6 (0.16 g, 0.85 mmol); the solution turned from bright yellow to dark green; a dark yellow solid was obtained; yield 64% (0.07 g, 0.11 mmol). Elemental analysis calc. for C19H23F6N4O3PRu %C: 37.94, %H: 3.85, %N: 9.32; found %C: 37.59, %H: 3.59, %N: 10.09. HR-MS: calc for C19H21N4O2Ru [M]+ m/z 439.0708, found m/z 439.011. 1H NMR (D2O, 500 MHz) δH: 1.02 (6H, d, J = 6.90), 2.08 (3H, s), 2.58 (1H, sep, J = 6.90), 6.04 (2H, d, J = 6.49), 6.29 (2H, d, J = 6.49), 7.66 (1H, s), 7.92-7.94 (2H, m), 9.19 (2H, dd, J = 1.97, J = 4.88), 9.90 (2H, dd, J = 1.99, J = 5.80).
Aqueous Solution Chemistry of the RuII Arene Formato Complex
The following experiment was carried out under normal ambient light conditions. Hydrolysis of the RuII arene formato complex was monitored by UV-vis spectroscopy. The nature of the hydrolysis products as well as the extent of the reaction were verified by 1H NMR spectroscopy or HR-MS. For UV-vis spectroscopy, the RuII arene formato complex was dissolved in H2O to give a 100 μM solution. The absorbance was recorded at several time intervals at the selected wavelength (at which the maximum changes in absorbance were registered) at 310 K over 8 h. A plot of the change in absorbance with time was computer-fitted to the pseudo first-order rate equation: A = C0 + C1e -kt (where C0 and C1 are computer-fitted constants and A is the absorbance corresponding to time) using Origin version 8.0 (Microcal Software Ltd.) to give the half-life (t1/2, min) and rate constant value (k, min-1). For 1H NMR spectroscopy, the RuII arene formato complex was dissolved in 90% H2O/10% D2O to give a 100 μM solution. The 1H NMR spectra at 310 K were recorded at various time intervals. The relative amounts of RuII arene formato species or aqua adduct were quantified (determined by integration of peaks in 1H NMR spectra).
Regioselective Reduction of NAD+ by RuII Arene Complexes in the Presence of Formate. The following experiment was carried out under normal ambient light conditions An equimolar amount of NAD+ was added to an NMR tube containing a 3.0 mM solution of the RuII arene halido complexes and an excess of NaHCO2 (25 mol equiv) in 90% H2O/10% D2O at ambient temperature. The 1H NMR spectra of the resulting solutions were recorded at 310 K at various time intervals for 24-48 h.
Regioselective Reduction of NAD+ by RuII Arene Complexes in the Presence of Formate upon UVA Photoirradiation. An equimolar amount of NAD+ was added to an NMR tube containing a 3.0 mM solution of the RuII arene pyridine complex and an excess of NaHCO2 (25 mol equiv) in 90% H2O/10% D2O at ambient temperature. 1H NMR spectra of the resulting solutions were acquired at different stages of photoirradiation with UVA (λirr = 300-400 nm) at 310 K for 12 h.
Acknowledgements
We thank the WPRS and the ORSAS and the CONACyT Mexico (S.B-L.) for research scholarships, the EPSRC, the ERC (BIOINCMED, grant no 247450), and ERDF/ AWM (Science City) for funding. Dr. Ivan Prokes, Dr. Lijiang Song, and Mr Philip Aston (University of Warwick) are acknowledged for assistance with the NMR and MS instruments, respectively.
References
1. Carrion, M. C.; Sepulveda, F.; Jalon, F. A.; Manzano, B. R.; Rodriguez, A. M. Organometallics 2009, 28, 3822-3833. [ Links ]
2. Sandoval, C. A.; Bie, F.; Matsuoka, A.; Yamaguchi, Y.; Naka, H.; Li, Y.; Kato, K.; Utsumi, N.; Tsutsumi, K.; Ohkuma, T.; Murata, K.; Noyori, R. Chem. Asian J. 2010, 5, 806-816. [ Links ]
3. Chaplin, A. B.; Dyson, P. J. Organometallics 2007, 26, 4357-4360. [ Links ]
4. (a) Canivet, J.; Süss-Fink, G. Green Chem., 2007, 9, 391-397; [ Links ] (b) Romain, C.; Gaillard, S.; Elmkaddem, M. K.; Toupet, L.; Fischmeister, Thomas, C. M.; Renaud, J.-L. Organometallics 2010, 29, 1992-1995. [ Links ]
5. (a) Ogo, S.; Makihara, N.; Watanabe, Y. Organometallics 1999, 18, 5470-5474; [ Links ] (b) Ogo, S.; Uehara, K.; Abura, T.; Watanabe, Y.; Fukuzumi, S. Organometallics, 2004, 23, 3047-3052; [ Links ] (c) Ogo, S.; Abura, T.; Watanabe, Y. Organometallics 2002, 21, 2964-2969. [ Links ]
6. Yan, Y. K.; Melchart, M.; Habtemariam, A.; Peacock, A. F. A.; Sadler, P. J. J. Biol. Inorg. Chem. 2006, 11, 483-488 [ Links ]
7. Herrmann, W. A; Kohlpaintner, C. W. Angew. Chem., Int. Ed. 1993, 32, 1524-1544. [ Links ]
8. (a) Cornils, B.; Hermann, W. A. Aqueous-Phase Organometallic Catalysis, 2nd ed., Wiley-VCH, 2002; [ Links ] (b) Lindstrom, U. M. Chem. Rev. 2002, 102, 2751-2772. [ Links ]
9. Himeda, Y.; Onozawa-Komatsuzaki, N.; Sugihara, H.; Arakawa, H.; Kasuga, K. J. Mol. Cat. A-Chem. 2003, 195, 95-100. [ Links ]
10. (a) Casey, C. P.; Bikzhanova, G. A.; Cui, Q.; Guzei, I. A. J. Am. Chem. Soc., 2005, 127, 14062-14071; [ Links ] (b) Zanotti-Gerosa, A.; Hems, W.; Groarke, M.; Hancock, F. Platinum Metals Rev. 2005, 49, 158-165. [ Links ]
11. Soleimannejad, J.; Sisson, A.; White, C. Inorg. Chim. Acta 2003, 352, 121-128 [ Links ]
12. Davenport, A. J.; Davies, D. L.; Fawcett, J.; Russell, D. R. Dalton Trans. 2004, 1481-1492. [ Links ]
13. Canivet, J., Karmazin-Brelot, L.; Süss-Fink, G., J. Organomet. Chem. 2005, 690, 3202-3211. [ Links ]
14. Bassetti, M.; Centola, F.; Sémeril, D.; Bruneau, C.; Dixneuf, P. H. Organometallics 2003, 22, 4459-4466. [ Links ]
15. Jan, D.; Delaude, L.; Simal, F.; Demonceau, A.; Noels, A. F. J. Organomet. Chem. 2000, 606, 55-64. [ Links ]
16. (a) Lozano-Vila, A. M.; Monsaert, S.; Bajek, A.; Verpoort, F. Chem. Rev., 2010, 110, 4865-4909; [ Links ] (b) Ung, T.; Hejl, A.; Grubbs, R. H.; Schrodi, Y. Organometallics 2004, 23, 5399-5401; [ Links ] (c) Peris, E.; Crabtree, R. H. Coord. Chem. Rev. 2004, 248, 2239-2246. [ Links ]
17. (a) Grundwald, C.; Gevert, O.; Wolf, J.; Gonzales-Herrero, P.; Werner, H. Organometallics 1996, 15, 1960-1962; [ Links ] (b) Drouin, S. D.; Yap, G. P. A.; Fogg, D. E. Inorg. Chem. 2000, 39, 5412-5414; [ Links ] (c) Nguyen, S. T.; Johnson, L. K.; Grubbs, R.; Ziller, J. W. J. Am. Chem. Soc. 1992, 114, 3974-3975. [ Links ]
18. (a) Lee, H. M.; Bianchini, C.; Jia, G.; Barbaro, P. Organometallics 1999, 18, 1961-1966; [ Links ] (b) Toledo, J. C.; Neto, B. S. L.; Franco, D. W. Coord. Chem. Rev., 2005, 249, 419-431; [ Links ] (c) Grela, K.; Harutyunyan, S.; Michrowska, A. Angew. Chem. Int. Ed. 2002, 41, 4038-4040. [ Links ]
19. Steckhan, E.; Herrmann, S.; Ruppert, R.; Dietz, E.; Frede, M.; Spika, E. Organometallics 1991, 10, 1568-1577. [ Links ]
20. Lo, H. C.; Leiva, C.; Buriez, O.; Kerr, J. B.; Olmstead, M. M.; Fish, R. H. Inorg. Chem. 2001, 40, 6705-6716. [ Links ]
21. Westerhausen, D.; Herrmann, S.; Hummel, W.; Steckhan, E. Angew. Chem., Int. Ed. Engl. 1992, 31, 1529-1531. [ Links ]
22. Betanzos-Lara, S.; Habtemariam, A.; Novakova, O.; Deeth, R. J.; Pizarro, A. M.; Clarkson, G. J.; Liskova, B.; Brabec, V.; Sadler, P. J., J. Biol. Inorg. Chem. 2012, DOI: 10.1007/s00775-012-0917-9. [ Links ]
23. Betanzos-Lara, S.; Salassa, L.; Habtemariam, A.; Nováková, O.; Pizarro, A. M.; Clarkson, G. J.; Liskova, B.; Brabec, V.; Sadler, P. J. Organometallics 2012, 31, 3466-3479. [ Links ]
24. Koike, T.; Ikariya, T. Adv. Synth. Catal., 2004, 346, 37-41. [ Links ]
25. (a) Crabtree, R. H.; Eisenstein, O.; Sini, G.; Peris, E. J. Organomet. Chem. 1998, 567, 7-11; [ Links ] (b) Peris, E. Jr.; Lee, J. C.; Crabtree, R. H. J. Chem. Soc. Chem. Commun. 1994, 2573-2574. [ Links ]
26. (a) Abdur-Rashid, K.; Clapham, S. E.; Hadzovic, A. J.; Harvey, N.; Lough, A. J.; Morris, R. H. J. Am. Chem. Soc. 2002, 124, 15104-15118; [ Links ] (b) Fryzuk, M. D.; MacNeil, P. A.; Rettig, S. J. J. Am. Chem. Soc. 1987, 109, 2803-2812. [ Links ]
27. Hayashi, H.; Ogo, S.; Abura, T.; Fukuzumi, S. J. Am. Chem. Soc. 2003, 125, 14266-14267. [ Links ]
28. Whittlesey, M. K.; Perutz, R. N.; Moore, M. H. Organometallics 1996, 15, 5166-5169. [ Links ]
29. de los Ríos, I.; Jiménez Tenorio, M.; Jiménez Tenorio, M. A.; Puerta, M. C.; Valerga, P. J. Organomet. Chem. 1996, 525, 57-64. [ Links ]
30. Betanzos-Lara, S.; Liu, Z.; Habtemariam, A.; Sadler, P. J., Angew. Chem. Int. Ed. 2012, 51, 3897-3900. [ Links ]
31. Vitinius, U.; Schaffner, K.; Demuth, M.; Heibel, M.; Selbach, H. Chem. Biodiv. 2004, 1, 1487-1497. [ Links ]
32. Fernández, R.; Melchart, M.; Habtemariam, A.; Parsons, S.; Sadler, P. J. Chem. Eur. J. 2004, 10, 5173-5179. [ Links ]
33. Govindaswamy, P.; Canivet, J.; Therrien, B.; Süss-Fink, G.; Štĕpnička, P.; Ludvík, J. J. Organomet. Chem. 2007, 692, 3664-3675. [ Links ]
34. Wang, F.; Habtemariam, A.; van der Geer, E. P. L.; Fernández, R.; Melchart, M.; Deeth, R. J.; Aird, R.; Guichard, S.; Fabbiani, F. P. A.; Lozano-Casal, P.; Oswald, I. D. H.; Jodrell, D. I.; Parsons, S.; Sadler, P. J. Proc. Natl. Acad. Sci. U.S.A. 2005, 102, 18269-18274. [ Links ]

