










Services on Demand
Journal
Article
 English (pdf)
English (pdf)
 Article in xml format
Article in xml format Article references
Article references
 Send this article by e-mail
Send this article by e-mailIndicators
-
 Cited by SciELO
Cited by SciELO -
 Access statistics
Access statistics
Related links
-
 Similars in
SciELO
Similars in
SciELO
Share

 Permalink
PermalinkJournal of the Mexican Chemical Society
Print version ISSN 1870-249X
J. Mex. Chem. Soc vol.57 n.2 Ciudad de México Apr./Jun. 2013
Article
Explicit Particle-number Dependence in Density Functional Theory
Samuel B. Trickey1* and Alberto Vela2*
1 Quantum Theory Project, Dept. of Physics and Dept. of Chemistry, P.O. Box 118435, University of Florida, Gainesville FL 32611-8435, USA. trickey@qtp.ufl.edu
2 Departamento de Química, Cinvestav, Av. IPN 2508, Colonia San Pedro Zacatenco México, D. F. 07360 México. avela@cinvestav.mx
Received July 9, 2012
Accepted April 4, 2013
Abstract
With rare exceptions, explicit particle number dependence (Ne-dependence) in approximate density functionals is viewed as a serious deficiency because of apparent size-consistency issues. In contrast, there are multiple manifestations of explicit Ne-dependence in density functional bounds (including the Gázquez-Robles kinetic energy bound), constraints, and approximations. We argue that these constitute inescapable motivation for exploring Ne-dependent approximate functionals. Doing so would be consistent with a mostly-ignored result of Lieb about properties of the universal functional.
Key words: Density functional theory, Hohenberg-Kohn theory, Kohn-Sham theory, particle number dependence, size-consistency.
Resumen
Con contadas excepciones, en la teoría de funcionales de la densidad aproximada la dependencia explícita en el número de partículas se visualiza como una seria deficiencia debido a problemas aparentes asociados con la consistencia en tamaño. En contraste, existen múltiples manifestaciones de cotas (como la cota de Gázquez-Robles de la energía cinética), restricciones y aproximaciones de los funcionales de la densidad las cuales tienen una dependencia explícita en el número de partículas. En este trabajo discutimos que lo anterior es una motivación ineludible para explorar la búsqueda y construcción de funcionales aproximados con dependencia explícita en el número de partículas. Hacerlo será consistente con un resultado debido a Lieb, y muy frecuentemente ignorado, respecto a la estructura del funcional universal.
Palabras Clave: Teoría de funcionales de la densidad, teoría de Hohenberg-Kohn, teoría de Kohn-Sham, dependencia en el número de partículas, consistencia en tamaño.
Dedication and Apology
We have had the privilege and pleasure of professional discussion, research collaboration, and personal friendship with José Luis Gázquez for many years. Broadminded and concerned as always for the growth of his students, Prof. Gázquez sent AVA, as a graduate student, to Gainesville to attend one of the last QTP Winter Institutes. Thus the co-authors of this paper met. Sometime earlier than that, SBT had met Gázquez, but now many years later, the actual year is vague. In the odd randomness that seems to characterize many career paths, the three of us did not publish together until 2012! In fact, the co-authors of this paper first published together in 2009, more than 20 years after our first meeting. Both of us, however, have been witnesses to the many key roles played by Prof. Gázquez in the development of density functional theory as a major topic in Mexican theoretical chemistry and materials physics and in physical science research and education in Mexico more generally. And, in the last year, our long relationship suddenly has flowered in four papers [1-4], with more expected. It is therefore with admiration and affection that we present this exploratory paper on a possible new direction for development of approximate density functionals.
This paper was intended for the issue honoring Prof. Gázquez. An unfortunate misunderstanding on the part of the first author led to missing the deadline. We apologize.
Size-extensivity
In both many-body physics and quantum chemistry, there is a deep aversion to methods which have an explicit dependence on electron number Ne, because of possible violation of both size-extensivity and size-consistency. Size-consistency is the requirement that the energy of a system consisting of two non-interacting subsystems equals the sum of the energies of the isolated subsystems, i. e., E(AB) = E(A) + E(B). The related concept, size-extensivity, refers to the fact that a quantity scales proportionally to the particle number: the total energy is an example. The aversion to number dependence is both reasonable and understandable. Consider, for example, the understanding of configuration interaction methods. There have been years of emphasis in quantum chemistry on the importance of both size-consistency and extensivity [the literature is too large to cite in detail with fairness, but see, for example, Nooijen et al. [5], Bartlett and Musial [6] and the many references in both]. That emphasis has led, for example, to a clear understanding of the relationship between formal properties of a full-CI and of various approximations. Part of the basic appeal of coupled-cluster theory lies in its correctness in this regard at each cluster order: systematic approximations without number-dependent difficulties are assured.
That aversion to number-dependence is not so commonly discussed in density functional theory (DFT) [7-12] but it is there. Because the constrained-search formulation [13, 14] of DFT follows from the ordinary variation principle, the assumption is that the ground-state energy functional E[n] is size-extensive. (We denote the electron density as n(r).) Indeed, the very language of a "universal functional" fits with that assumption. Savin has discussed exceptions related to degeneracies [15]. In our experience, such exceptions generally are viewed as failures or dismissed as inconsequential oddities. Certainly, the dependences on the number of electrons and the degeneracy of the ground states are fundamental and related issues [16]. However, for the sake of simplicity, it may be helpful and useful to approach the former problem first, hoping that from its study we will find out more formal and rigorous ways to tackle the latter. Some steps in this direction have already been explored [17-19].
Here we propose that it is time to explore a different line altogether. In essence, our argument is that the long history of developing approximate exchange-correlation functionals Exc[n] and approximate Kohn-Sham (KS) kinetic density functionals Ts[n] includes multiple indications that it is appropriate to build approximate functionals which are explicitly Ne-dependent. The argument is strengthened by recognition that those practical indications are consistent with a known but generally ignored basic exact DFT result. We hasten to add that this is not a call to violate, somehow, thermodynamic limits. We include a brief discussion of that issue.
Number-dependence in Density-functional Theory
Approximate Functionals and Bounds
To set notation and units, we display non-spin-polarized Kohn-Sham [KS] DFT in its usual form. In hartree atomic units, the total energy functional is


with n(r) the electron number density, nj the occupation numbers, and vext(r) the external potential (ordinarily, the nuclear-electron Coulomb interaction). In order, Eee is the Hartree or classical electron-electron Coulomb repulsion energy, Exc is the DFT exchange-correlation energy (which comprises both Coulombic and kinetic energy contributions), and Eext is the external interaction energy. The orbitals ji are eigenstates of the KS Hamiltonian, details of which are of no consequence here.
One of the earliest manifestations, perhaps the first, of explicit number dependence in a many-electron energy was the Fermi-Amaldi correction [20] to the Thomas-Fermi energy
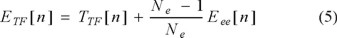
The explicit form of the TF kinetic energy is of no concern here. What is relevant is the prefactor to Eee. It is designed to remove, at least in an average sense, the spurious self-interaction error in the Hartree energy. Interestingly, one of José Luis Gázquez' earlier papers is "An exchange energy functional based on the Dirac and the Fermi-Amaldi approximations" [21]. That paper presents a simple, orbital-independent but Ne-dependent modification of the Dirac-Slater exchange functional which, by virtue of the Fermi-Amaldi correction, is one-electron self-interaction free, to wit:

The constants (C1xFA = -0.7386, C2xFA = 1.000; subscript "xFA" for "exchange-Fermi-Amaldi") were set by a combination of simple consistency arguments and least-squares fitting to the Hartree-Fock exchange energies of a large sample (1785) of atoms and ions. Those values are not critical to the present discussion. What is critical (but not entirely surprising) is that the highest occupied KS eigenvalue from this simple Ne-dependent representation comes much closer to respecting the DFT IP theorem [22] than does the eigenvalue of simple Dirac-Slater-exchange. Usually such improvement is obtained by introducing exact exchange [12, 23] either as a global [24, 25] or a range-separated hybrid functional [26, 27]. We remark, in passing, that in Ref. [28] Ayers et al. discuss size inconsistency of the Fermi-Amaldi model, the F-A correction due to Silver and Bartlett [29], and the Parr-Ghosh corrected F-A procedure [30].
An inequality which has been used (and debated) to constrain approximate exchange functionals, e.g. PW91 [31], PBE [32, 33] and our own VMT [34] and VT{8,4} [3], is the Lieb-Oxford bound [35, 36]. The form commonly used in this context is

The conventional value of the LO constant is λLO = 2.273, or the slightly tighter Chan-Handy value [37], λCH = 2.215. Again, the difference is not relevant to the present discussion.
With few exceptions, it generally is not noticed that the LO bound, in fact, depends on Ne [35-37] and the dimensionality of space [38-40]. As discussed by Odashima et al. [41], several facts are known about that Ne-dependence. For Ne = 1, λ(1) = 1.48, while for Ne = 2, λ(2) ≥ 1.67. Moreover, for all Ne , λ(Ne + 1) ≥ λ(Ne). Ref. [41] explored the effect of using these facts in the most obvious way to re-parameterize the PBE X functional. The device used was a simple smooth interpolation of the form

When enforced on PBE X, there was little, if any, improvement in performance. In view of the derivative discontinuity in the universal functional at integer Ne [42], it may be that the smooth interpolation, Eq. (9), masks some important features. Or it may be simply that the PBE X functional form is insensitive to details of the enforcement of the LO bound. Whatever the cause, that does not vitiate the underlying fact of the number dependence of the LO bound.
For several years, SBT has been quite involved in developing orbital-free kinetic energy functionals as approximations to the explicitly orbital-dependent form of Ts[n] defined in Eq. (2). More recently AVA has become involved in this quest. See Refs. [43-45] for example. For the construction of such approximate functionals, it is valuable to consider another functional which bounds Ts[n], i.e. some Tbound[n] ≥ Ts[n]. Stated more generally, suppose an Ne fermion state Ψ, with associated number density nΨ. Then the desired functional is

In 1982, Gázquez and Robles [46] gave the following bound on the KS KE for each spin (labeled σ= α, β):
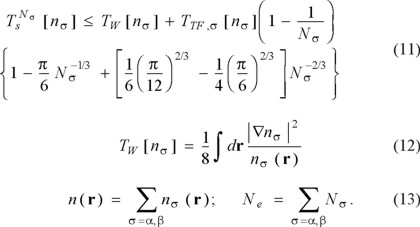
Although Gázquez and Robles wrote this as a density functional (i.e., an approximation for Ts), we write it as a bound because they omitted terms of higher order in Ne -1/3. Again, the explicit number dependence is the central point for the present discussion.
Zumbach [47] has given a somewhat similar bound on the KS kinetic energy, to wit:
Ts[n] ≤ (1 + CZNe2/3)TW (14a)
CZ = 9(4π)2(2/5)2/3. (14b)
(Subscript "Z" for Zumbach; beware that his paper is in Rydberg au.) Again, an explicit number dependence appears.
The bound given by Gázquez and Robles, Eq. (11), was motivated, at least in part, by a semi-empirical procedure of Acharya et al. [48] which yielded an approximate Ts[n] of the form
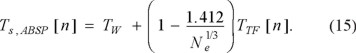
(Subscript ABSP for the authors of that paper.) For singlets with N= 1, 2, this must reduce to Ts,ABSP = TW, so the Acharya et al. expression is valid for Ne ≥ 3. Many other bounds of similar character, with references to the original literature, are collected in the review by Ludeña and Karasiev [49].
From the perspective of developing approximate KS KE functionals, Eqs. (11), (14a), and (15) all are examples of the theorem on the positivity of the so-called Pauli term Tϑ [50-53]
Ts[n] = TW[n] + Tθ[n], Tθ[n] ≥ 0 (16)
in the exact decomposition of the KS KE. But the same argument applies to this exact decomposition as to the Acharya et al. [48] approximation. For Ne = 1 and a singlet with Ne = 2, Tϑ must vanish, yet for an Ne = 2 triplet that is not true. This is an extremely subtle and difficult Ne-dependence to reproduce with an approximate functional which is not explicitly Ne-dependent.
An informative discussion of this subtlety was given by Chan and Handy [54]. They pointed out that the Lieb-Thirring bound [55]
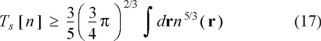
is violated by TW[n] for sufficiently large n. That in itself is enough to establish Tϑ[n] ≥ 0. Moreover, they in essence established the Ne-dependence of Tϑ[n] by using density scaling arguments, n(r) → λn(r), to establish that Ts[n] is not homogeneous of degree 1. Density scaling is, by design, number-changing and, as such, it reveals the derivative discontinuities at integer Ne.
Recently Alipour and Mohajeri [56] have given a new information-theoretic determination of the kinetic energy contribution Tc to the DFT exchange-correlation energy. Both the information energy and Shannon entropy forms from which they obtain fitted expressions are explicitly Ne-dependent.
To conclude this survey, note the recent interest in tuning [57] the separation parameter in range-separated hybrids to enforce the exact behavior of the IP theorem, namely that for the exact ground-state functional, the IP is the negative of the HOMO KS eigenvalue, εHOMO = -IP. This tuning is done specifically for each system under study, hence is manifestly Ne-dependent.
Exact functionals
We have cataloged and discussed these examples of explicit and implicit Ne-dependence from the perspective of developers of approximate density functionals. But there is more to the issue than forms of approximate functionals and constraints on them. We note three examples and a general case.
Derivative discontinuities [42] play a key role in the definition and use of hardness and softness (another of JLG's interests [58-60]). This is true both for the exact definitions and for approximate functionals which interpolate what should be linear relations, yet another of JLG's interests. See discussions and references in Ayers [61] and in Cohen et al. [23] The obvious relevance to this discussion is that those discontinuities occur at integer Ne. Relative to a specified Ne, the slope on the electron-deficient side of E[N] gives the IP (with the EA on the other side). One can infer from the IP theorem that the functional at Ne involves information about E[Ne-1] therefore.
Second, we note that exact exchange in DFT is intrinsically Ne-dependent. The functional itself sums over Ne orbitals. This number dependence is not as easy to hide as in the usual KS case, in which Ts is the only manifestly Ne-dependent contribution. One can see this from the simple fact that Ts and Ex scale differently under uniform density scaling; recall the discussion above about the Chan-Handy results.
The third exact result is the expression derived by Levy, Perdew and Sahni [22] for the exact effective potential in orbital-free DFT

Here µ is the negative of the ionization potential. They obtain this result by factoring the exact, normalized, Ne-electron ground-state wave function into
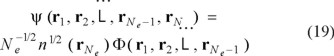
From that they obtain a positive-definite expression for veff explicitly in terms of the conditional distribution F(r1,r2,...,rNe-1), which itself is explicitly number dependent.
All of these examples illustrate the remark by Lieb [14] (at the outset of his section 4.A) that any mathematically satisfactory definition of the functional (1) "... must depend explicitly on the particle number Ne. This fact is unavoidable and frequently overlooked." We add that the remark itself is almost completely overlooked. The only exception that comes to mind is the discussion by Ludeña and Karasiev in the volume in honor of R.G. Parr. [49] After surveying various approximate Ne-dependent forms for Ts (p. 624), they conclude on p. 659 with "Finally, an important challenge arises from the fact that exact treatments such as those based on the solution of a differential equation TS, or on local-scaling transformations lead to an Ne-dependent enhancement factor. This finding should be investigated as it might help us to understand the puzzling question regarding whether or not universal functionals really exist."
A simplified version of the argument for mandatory explicit Ne-dependence of the exact DFT functional comes from the realization that the first Hohenberg-Kohn theorem implies that the ground-state energy is a function of Ne and a functional of the external potential vext(r)
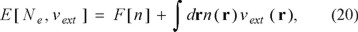
where the notation for the universal functional F[n] indicates that the only explicit dependence in this functional is on the electron density, whereas the total energy and the external energy depend, in general, on both the choice of external potential and Ne, If the external potential is changed by dvext(r), the resulting energy first-order energy change is

and the electron density and the external potential thus are conjugate variables, like velocity and momentum, since they are related by

In essence, this conjugation allows us to consider inverse Legendre transform DFT, in which the dependence on the external potential is replaced by its conjugate variable, the electron density. The corresponding transform is given by
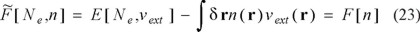
where the last equality is obtained after substituting Eq. (20) in the Legendre transform. Eq. (23) simply indicates that  and F are the same mathematical object but the new piece of information that we have gained is that indeed, the universal functional has an explicit dependence on the number of electrons. The normalization condition can be used to eliminate the explicit dependence on Ne, i. e., [Ne,n] = F[Ne[n],n] = F[n], leaving a functional that only depends on the electron density.
and F are the same mathematical object but the new piece of information that we have gained is that indeed, the universal functional has an explicit dependence on the number of electrons. The normalization condition can be used to eliminate the explicit dependence on Ne, i. e., [Ne,n] = F[Ne[n],n] = F[n], leaving a functional that only depends on the electron density.
Pursuing Explicitly Number-dependent Approximate Functionals
Except for the tuning of range-separated hybrids mentioned above, common practice is to ignore the "unavoidable fact" about Ne-dependence in the development of approximate functionals. Another major exception, though it is not stated in such terms, is hybrid XC functionals used directly (without an optimized effective potential, i.e. a so-called generalized KS calculation). Such functionals have a contribution from explicit single-determinant exchange, which is Ne-dependent. Our emphasis is different. From the evidence summarized above, however, it seems clear that there are many imperatives for exploring the construction of Ne-dependent functionals. Before brief comment on the advantages of doing so, we must return to the size-consistency question.
Observe that total particle number in a system, indeed the definition of what constitutes the system, is a matter of quantum-mechanical preparation. Hence a diatomic molecule with Ne electrons separates into two atoms A, B with NeA + NeB = Ne but the atoms still are part of the same system. Prepared as a molecule, it remains a molecule (albeit one with zero binding energy) even at arbitrarily large separation, not two isolated atoms. From this perspective, size consistency is a matter of QM preparation: a separated molecule with Ne electrons is not the same QM preparation as multiple isolated atoms with the same nuclei and same total electron number.
An example of the distinction and its invocation in actual computation with an approximate functional is in Ref. [41], in particular, the discussion associated with its Table 1. The specific case was the Li2 molecule treated with the PBE X functional modified such that the LO bound is set by Eq. (9). The comparison was with PBE X using two standard estimates and one conjectured estimate of the Ne→∞value of λ. Straightforward use of the Ne-dependent functional to calculate diatomic molecule atomization energies ΔEled to nonsense, in that the Ne-dependent versions gave 50% greater ΔEthan the Ne→∞versions. In contrast, the calculated equilibrium bond lengths Re varied quite sensibly among the models, by about 0.01 Å at most. The difficulty was naïve violation of size-consistency, namely an atomization energy calculated from
ΔE = ELi2[λ(Ne = 6)] -2ELi[λ(Ne = 3)]. (24)
Clearly this prescription is size-inconsistent. There is no corresponding inconsistency in the Re values because they are not a difference between a molecular property and an atomic one. The inconsistency is one of quantum mechanical preparation. The properly prepared system uses the molecular particle number throughout, hence replaces (24) with
ΔE = ELi2[λ(Ne = 6)] -2ELi[λ(Ne = 6)]. (25)
At the risk of pedantry, we emphasize that the atomic total energy in (25), ELi [λ(Ne = 6)], is not the isolated atom total energy ELi[λ(Ne = 3)]. Adoption of an Ne-dependent functional inevitably means that the separated atom limit of the energies of an aggregated system is not the same as the sum of the isolated atom energies.
For a single truly finite system (molecule, cluster), generalization of the procedure in (25) is fairly straightforward: aggregate quantities are to be compared with separated-atom quantities calculated with the Ne value of the aggregate. Along a reaction path, matters get more complicated. After the fact, one could pick the largest aggregate along the path to set Ne, but that runs the risk of being circular reasoning. The preferred intermediates depend upon energetics, hence upon the selected Ne. Generalization of the protocol also is not so obvious for periodically bounded systems. In basic DFT applied to periodic systems, the Ne value is set to the unit cell. But a unit cell, of course, can be larger than the primitive unit cell for a given crystal. At present, our thinking is that the primitive unit cell of a given crystal should be normative for setting Ne. Again, the difficulty is in systematic comparison of aggregates. An example is a pressure-induced transition from fcc to hcp (one atom per primitive cell to two) in an elemental solid with atomic number Z. Straightforward extension of the finite-aggregate rule would have one use Ne = 2Z, but we do not know at this point whether that is sensible or leads to some unpleasant, unphysical behavior.
Closing Remarks
Finally, we make a few remarks about advantages of explicitly electron-number-dependent approximate density functionals. From the perspective of orbital-free DFT, Eqs. (11) and (15) show one clear advantage. The designer of an approximate KS KE functional can achieve satisfaction of (16) for Ne = 1,2, as well as the Thomas-Fermi limit for arbitrarily large particle number, much more easily than with an approximate functional without that explicit Ne-dependence. For the improved satisfaction of the DFT IP theorem, the tuned range-separated hybrid functionals already show the value of Ne-dependence. Perhaps the largest advantage, however, is that of opening a new route for progress. Though it may seem uncomfortably speculative, opting to exploit an unavoidable aspect of theory may eventually prove to be more productive than continuing the struggle to ignore that aspect in the construction of approximate functionals.
Acknowledgements
AV was supported in part by the Conacyt project grant 128369. SBT was supported in part by the U.S Dept. of Energy grant DESC-0002139. He thanks G.G. Emch for a trenchant and helpful clarification of the issue of quantum mechanical preparation.
References
1. del Campo, J. M.; Gázquez, J. L.; Trickey, S. B.; Vela, A. J. Chem. Phys. 2012, 136, 104108. [ Links ]
2. Gázquez, J. L.; del Campo, J. M.; Trickey, S. B.; Álvarez-Méndez, R. J.; Vela, A. In: "Concepts and Methods in Modern Theoretical Chem-istry, Vol. 1", in honor of Professor B.M. Deb, edited by S. K. Ghosh and P. K. Chattaraj, 2013, p. 295. [ Links ]
3. Vela, A.; Pacheco-Kato, J. C.; Gázquez, J. L.; del Campo, J. M.; Trickey, S. B. J. Chem. Phys. 2012, 136, 144115. [ Links ]
4. del Campo, J. M.; Gázquez, J. L.; Trickey, S. B.; Vela, A. Chem. Phys. Lett. 2012, 543, 179. [ Links ]
5. Nooijen, M.; Shamasundar, K. R.; Mukherjee, D. Mol. Phys. 2005, 103, 2277. [ Links ]
6. Bartlett, R. J.; Musial, M. Rev. Mod. Phys. 2007, 79, 291. [ Links ]
7. Kohn, W.; Sham, L. J. Phys. Rev. 1965, 140, A1133. [ Links ]
8. Parr, R. G.; Yang, W. T. Density Functional Theory of Atoms and Molecules, Oxford University Press, New York 1989. [ Links ]
9. Dreizler, R. M.; Gross, E. K. U. Density Functional Theory: an approach to the Quantum Many-Body Problem, Springer-Verlag, Berlin, Heidelberg 1990. [ Links ]
10. Fiolhais, C.; Nogueira, F.; Marques, M. A. L. A Primer in Density Functional Theory, Springer, Berlin 2003. [ Links ]
11. Perdew, J. P.; Ruzsinszky, A.; Tao, J. M.; Staroverov, V. N.; Scuseria, G. E.; Csonka, G. I. J. Chem. Phys. 2005, 123, 062201. [ Links ]
12. Scuseria, G. E.; Staroverov, V. N. In: Theory and Applications of Computational Chemistry: The First Forty Years, edited by C. E. Dykstra, G. Frenking, K. S. Kim and G. E. Scuseria. Elsevier, Amsterdam, 2005, p. 669. [ Links ]
13. Levy, M. Proc. Natl. Acad. Sci. USA 1979, 76, 6062. [ Links ]
14. Lieb, E. H. Int. J. Quantum Chem. 1983, 24, 243. [ Links ]
15. Savin, A. Chem. Phys. 2009, 356, 91. [ Links ]
16. Yang, W. T.; Zhang, Y. K.; Ayers, P. W. Phys. Rev. Lett. 2000, 84, 5172. [ Links ]
17. Ayers, P. W. Theor. Chem. Acc. 2007, 118, 371. [ Links ]
18. Gál, T. Phys. Rev. A 2001, 63, 022506. [ Links ]
19. Gál, T.; Geerlings, P. Phys. Rev. A 81, 2010, 032512. [ Links ]
20. Fermi, E.; Amaldi, E. Accad. Ital. Rome 1934, 6, 117. [ Links ]
21. Cedillo, A.; Ortiz, E.; Gázquez, J. L.; Robles, J. J. Chem. Phys. 1986, 85, 7188. [ Links ]
22. Levy, M.; Perdew, J. P.; Sahni, V. Phys. Rev. A 1984, 30, 2745. [ Links ]
23. Cohen, A. J.; Mori-Sánchez, P.; Yang, W. Chem. Rev. 2012, 112, 289. [ Links ]
24. Becke, A. D. J. Chem. Phys. 1993, 98, 5648. [ Links ]
25. Becke, A. D. J. Chem. Phys. 1993, 98, 1372. [ Links ]
26. Leininger, T.; Stoll, H.; Werner, H. J.; Savin, A. Chem. Phys. Lett. 1997, 275, 151. [ Links ]
27. Toulouse, J.; Colonna, F.; Savin, A. Phys. Rev. A 2004, 70, 062505. [ Links ]
28. Ayers, P. W.; Morrison, R. C.; Parr, R. G. Mol. Phys. 2005, 103, 2061. [ Links ]
29. Silver, D. M.; Bartlett, R. J. Phys. Rev. A 1976, 13, 1. [ Links ]
30. Parr, R. G. Ghosh, S. K. Phys. Rev. A 1995, 51, 3564. [ Links ]
31. Perdew, J. P. In: Electronic Structure of Solids '91, edited by P. Ziesche and H.Eschrig, Akademie Verlag, 1991, p. 11. [ Links ]
32. Perdew, J. P.; Burke, K.; Ernzerhof, M. Phys. Rev. Lett. 1996, 77, 3865. [ Links ]
33. Perdew, J. P.; Burke, K.; Ernzerhof, M. Phys. Rev. Lett. 1997, 78, 1396. [ Links ]
34. Vela, A.; Medel, V.; Trickey, S. B. J. Chem. Phys. 2009, 130, 244103. [ Links ]
35. Lieb, E. H. Phys. Lett. A 1979, 70, 444. [ Links ]
36. Lieb, E. H.; Oxford, S. Int. J. Quantum Chem. 1981, 19, 427. [ Links ]
37. Chan, G. K. L.; Handy, N. C. Phys. Rev. A 1999, 59, 3075. [ Links ]
38. Rasanen, E.; Pittalis, S.; Capelle, K.; Proetto, C. R. Phys. Rev. Lett. 2009, 102, 206406. [ Links ]
39. Rasanen, E.; Pittalis, S.; Proetto, C. R.; Capelle, K. Physica E 2010, 42, 1236. [ Links ]
40. Rasanen, E.; Seidl, M.; Gori-Giorgi, P. Phys. Rev. B 2011, 83, 195111. [ Links ]
41. Odashima, M. M.; Capelle, K.; Trickey, S. B. J. Chem. Theory Comput. 2009, 5, 798. [ Links ]
42. Perdew, J. P.; Parr, R. G.; Levy, M.; Balduz, J. L. Phys. Rev. Lett. 1982, 49, 1691. [ Links ]
43. Karasiev, V. V.; Jones, R. S.; Trickey, S. B.; Harris, F. E. Phys. Rev. B 2009, 80, 245120. [ Links ]
44. Dufty, J. W.; Trickey, S. B. Phys. Rev. B 2011, 84, 125118. [ Links ]
45. Trickey, S. B.; Karasiev, V. V.; Vela, A. Phys. Rev. B 2011, 84, 075146. [ Links ]
46. Gázquez, J. L.; Robles, J. J. Chem. Phys. 1982, 76, 1467. [ Links ]
47. Zumbach, G.; Phys. Rev. A 1985, 31, 1922. [ Links ]
48. Acharya, P. K.; Bartolotti, L. J.; Sears, S. B.; Parr, R. G. Proc. Natl. Acad. Sci. USA 1980, 77, 6978. [ Links ]
49. Ludeña, E.; Karasiev, V. V. In: Rev. Mod. Quantum Chem. "A Celebration of the Life of R.G. Parr", edited by K. D. Sen. World Scientific, Singapore, 2002, p. 612. [ Links ]
50. Tal, Y.; Bader, R. F. W. Int. J. Quantum Chem. 1978, S12, 153. [ Links ]
51. Bartolotti, L. J.; Acharya, P. K. J. Chem. Phys. 1982, 77, 4576. [ Links ]
52. Harriman, J. E. In: Density Matrices and Density Functionals, edited by R. Erdahl and V. H. Smith. D. Reidel, Dordrecht, 1987, p. 359. [ Links ]
53. M. Levy and H. Ou-Yang, Phys. Rev. A 38, 625 (1988). [ Links ]
54. G. K. L. Chan and N. C. Handy, Phys. Rev. A 59, 2670 (1999). [ Links ]
55. E. H. Lieb and W. E. Thirring, Phys. Rev. Lett. 35, 687 (1975). [ Links ]
56. M. Alipour and A. Mohajeri, Mol. Phys. 109, 1967 (2011). [ Links ]
57. Baer, R.; Livshits, E.; Salzner, U. Annu. Rev. Phys. Chem. 2010, 61, 85. [ Links ]
58. Gázquez, J. L. In: Chemical Reactivity Theory: A Density Functional View, edited by P. K. Chattaraj. CRC Press, Boca Raton FL US, 2009, p. 7. [ Links ]
59. Gázquez, J. L.; Garza, J.; Hinojosa, F. D.; Vela, A. J. Chem. Phys. 2007, 126, 214105. [ Links ]
60. Gázquez, J. L.; Garza, J.; Vargas, R.; Vela, A. In: AIP Conference Proceedings 979 (Recent Developments in Physical Chemistry) 2008, p. 11. [ Links ]
61. Ayers, P. W.; J. Math. Chem. 2008, 43, 285. [ Links ]

