










Servicios Personalizados
Revista
Articulo
 Inglés (pdf)
Inglés (pdf)
 Artículo en XML
Artículo en XML Referencias del artículo
Referencias del artículo
 Enviar artículo por email
Enviar artículo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO -
 Accesos
Accesos
Links relacionados
-
 Similares en
SciELO
Similares en
SciELO
Compartir

 Permalink
PermalinkJournal of the Mexican Chemical Society
versión impresa ISSN 1870-249X
J. Mex. Chem. Soc vol.57 no.1 Ciudad de México ene./mar. 2013
Article
Substituent Effect in para Substituted Osmabenzene Complexes
Reza Ghiasi* and Allieh Boshak
Department of Chemistry, Basic Science Faculty, East Tehran Branch, Qiam Dasht, Tehran, Islamic Azad University, Tehran, Iran. rezaghiasi1353@yahoo.com, rghyasi@qdiau.ac.ir
Received November 2012.
Accepted January 16, 2013.
Abstract
The electronic structure and properties of the osmabenzene and para substituted osmabenzenes have been explored using the hybrid density functional mpw1pw91 theory. The substituent effects of F, CH3, OH, CN, NO2, CHO and COOH in para osmabenzenes complexes were studied. Basic measures of aromatic character were derived from the structure and nucleus-independent chemical shift (NICS). Quantum theory of atoms in molecules analysis (QTAIM) indicates a correlation between ρ(Os-C) bonds and the electron density of bond critical point in all species.
Key words: Osmabenzene, substituent effect, DFT calculations, quantum theory of atoms in molecules, nucleus-independent chemical shift.
Resumen
Las estructuras electrónicas y propiedades de osmabencenos y osmabencenos sustituidos en para han sido exploradas mediante el uso de la teoría de los funcionales de densidad con el funcional híbrido mpw1pw91. Se estudiaron los efectos del sustituyente en para-de osmabencenos. Las medidas básicas del carácter aromático se derivaron a partir de la estructura y del desplazamiento químico independiente del núcleo (DQIN). El análisis mediante la teoría cuántica de átomos en moléculas indica una correlación entre los enlaces ρ(Os-C) y la densidad electrónica del punto crítico de enlace en todas las especies.
Palabras clave: Osmabenceno, efecto del sustituyente, cálculos TFD, teoría cuántica de átomos en moléculas, desplazamiento químico independiente del núcleo.
Introduction
Metallabenzenes are six-membered metallacycles analogous to benzene for which one CH unit has been replaced by an isolobal transition-metal fragment {MLn} [1-3]. In the past decade, the synthesis of metallabenzenes has been studied [4-6]. The first metallabenzene to be isolated and characterized was osmabenzene Os(C5H4[S])(CO)(PPh3)2, the OsC5 ring was assembled from two ethyne molecules and a single carbon atom from the thiocarbonyl ligand already resident on the osmium in the starting material, Os(CS)(CO)(PPh3)3 [7, 8]. The structure and properties of osmabenzenes have been studied experimentally and theoretically [9-14]. From experimental and theoretical examinations one sees that the actual experimental knowledge concerning osmabenzenes compounds is still relatively limited due to the subtle nature of such compounds. In the present study, the geometries, aromaticity and chemical bonding of osmabenzenes and para substituted osmaabenzenes are studied theoretically. The analysis of the electron density within the AIM methodology has been used for providing valuable information on characterizing the molecules based on their critical points.
Computational Methods
All calculations were carried out with the Gaussian 2003 suite of program [15] using the standard 6-311G++(d,p) basis set calculations of systems contain C, H, N, F, Cl and O [16, 17]. For Os the standard LANL2DZ basis set [18-20] was used and this element is described by an effective core potential (ECP) of Wadt and Hay pseudopotential [21]. Geometry optimization was performed utilizing the one parameter hybrid functional based on a modified Perdew-Wang exchange and correlation (mpw1pw91) [22]. A vibrational analysis was performed at each stationary point found, confirming its identity as an energy minimum.
The nucleus independent chemical shift (NICS) was used as a descriptor of aromaticity from the magnetic point of view. This index is defined as the negative value of the absolute magnetic shielding computed at ring centers [23] or any another interesting point of the system [24]. Rings with highly negative values of NICS are quantified as aromatic by definition, whereas those with positive values are anti-aromatic. The AIM2000 program was used for topological analysis of electron density [25]. The following characteristics of ring critical points (RCPs) are taken into account: density at RCP (ρ(rc)) and its Laplacian (∇2(rc)).
Result and discussion
Energetic analysis
Absolute energies of substituted C5H4XOs(PH3)2(CO)Cl complexes have been calculated in the singlet and quintet ground states of all molecules (Table 1). The comparison of these values indicates the major stability for singlet ground state in all molecules. On the other hand, the ionization energy and electron affinity of all molecules have been calculated (Table 1). For the electron withdrawing substituents (these having large positive values of substituent constants), were found the larger ionization energy and electron affinity values. There is good linear correlation between the electron affinity values and Hammet constants of substitutions (ςp) (R2 = 0.929).
Bond distances
The selected bond distances of substituted C5H4XOs(PH3)2 (CO)Cl molecules are presented in Table 2. As it can be seen in Table 2, the Os–C bond length in substituted C5H4XOs(PH3)2 (CO)Cl is changing due to the presence of the substituent in para position of the osmabenzene ring (Fig. 1). The Os-C1 bond is longer in the strongest electron donating substituents rather than weaker electron donating substituents. Therefore, the electron withdrawing substituents additionally stabilize the Os-C bond, while the electron donating ones weaken it. In addition, the Os–C1 bonds are relatively longer than their OsC5 counterparts. This can be explained by the presence of the chlorine ligand, which, as a lower field ligand than the CO one, weakens the cis-placed Os–CO bonds.
On the other hand, donating electron subsituents decrease the Os-C(O) bonds length. The increasing of metal charge in the presence of electron donating substituents causes to increasing back bonding in Os-C(O) bond, while the electron donating ones increase it.
Also, structural analysis indicates that the Os-C1 bonds are shorter on doublet ground state of cation form. On the other hand, the Os-C5 bond distances increase in doublet ground state of cation form. As shown in Table 2, Os-C1 and Os-C5 bond distances increase in anion form.
Frontier orbital energies and chemical hardness
The frontier orbital energies, HOMO-LUMO gap energy, hardness, chemical potential, and electrophilicity of all complexes computed are given in the Table 2. The graphical representations of frontier orbitals are shown in Figure 2. The effect of substitutions on the HOMO and LUMO energies has been analyzed by plotting the values of their energies against Ham-met substituent constants (ς). Figure 3 shows that the σ values correlate linearly with HOMO and LUMO energies. As it can be seen, the LUMO orbital can be involved in ϖ-electron interaction with d-orbital of the transition metal. These values indicate the energy of the frontier orbitals is less in the case of electron-withdrawing substituents.
The hardness and chemical potential of these complexes calculated from the HOMO and LUMO orbital energies using the following approximate expression:
µ = (εHOMO + εLUMO)/2
η = (εHOMO − εLUMO)/2
Where µ is the chemical potential (the negative of the electronegativity), and η is the hardness [26, 27].
The hardness values in Table 3 indicate the increasing of these values in donating substituents. On the other hand, these values decrease with electron-withdrawing substituents. These values show that the σ values correlate linearly with chemical potential values (R2 = 0.812).
The chemical potentials were also evaluated for this set of molecules. The chemical potential characterizes the tendency of electrons to escape from the equilibrium system. The values of chemical potential show that they increase for donating substituents and decrease for electron-withdrawing substituents (Table 3). These values show that the σ values correlates linearly with chemical potential values (R2 = 0.959), i.e. the chemical potential increase with the inductive and resonance effects caused by the substitution.
To evaluate the electrophilicity of these complexes, we have calculated the electrophilicity index, ω, for each complex measured according to Parr, Szentpaly, and Liu [28] using the expression:
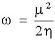
The values of electrophilicity index are shown in Table 3. It has been seen that the σ values correlate linearly with electrophilicity values (R2 = 0.914). These values show that the electrophilicity increases with the inductive and resonance effects caused by the substitution.
It is noticed that the Hammet correlation for chemical potential is better than for electrophiliity and hardness.
Nucleus-independent chemical shift analysis (NICS)
The nucleus-independent chemical shift (NICS) method has been widely employed to characterize aromaticity [29]. As an effort to discuss the use of NICS as a measure of aromaticity for six-membered rings, we have calculated NICS values along the z-axis to the ring plane beginning on the center of the ring up to 2.0 Å. These calculations show that the shape of the NICS pro-file with respect to the distance from the ring center is similar. In addition, for all species, we have localized the NICS maxima and minima and determined the distances to the center of the ring at which they occur. See Table 4. For all molecules, the highest absolute value of NICS is above the center of the ring. It is possible that induced magnetic fields generated by the ϖ aromaticity show a minimum NICS at the certain distance from the center of the ring. From Table 4, it can be seen that all six-membered ring compounds have large negative NICS values, indicating their enhanced aromatic properties. All these NICS values can be attributed to the delocalized π electrons current. Theoretical computation of all molecules show that there is a linear correlation between NICS (1.5) values and Hammet constants (Fig. 4). These values show that aromaticty increases with electron-withdrawing substituents.
QTAIM analysis
It has been proved, that the AIM-based analysis of electron density can provide valuable information on many physical and chemical properties of the molecular systems [30-37]. Two methods have been used for this analysis (Table 5). It has been found for instance that the value of electron density (ρ) and its Laplacian (∇2ρ) estimated at bond critical point (BCP) of a given bond correlate very well with the strength of this bond, as well as with its length, since, as it is well known, both the strength and the length of a given bond are mutually dependent [38-42]. Also in this case such a relationship can be observed and the linear regression can be found between ρ(Os-C) and r(Os-C) in all complexes (Fig. 5).
Additional valuable information on chemical bond properties is available from the total electron energy density, H(ρ), and its components; kinetic electron energy density, G(ρ), positive by definition, and potential electron energy density, V(ρ), negative by definition. The following relation is known for H(q) and its components [43,44]:
H(ρ) = V(ρ) + G(ρ)
It is known that in the region of the bond CP of weak closed-shell interatomic interactions the kinetic energy density dominates, with G(ρ) magnitude being slightly greater than the potential energy density |V(ρ)| which implies the total energy density H(ρ) > 0 and close to zero, whereas for strong covalent interactions V(ρ) dominates over the kinetic energy density and H(ρ) < 0. This is usually accompanied by ∇2ρ > 0 for the proper case and ∇2ρ < 0 for the latter one (there is one exception mentioned in the further part of the discussion).
Os-C bond
Interestingly, both G(ρ) and |V(ρ)| values increase with electron-withdrawing properties of substituents in the para-position of the osmabenzene ring. However, H(ρ) is invariably positive and very close to zero. Probably, this could be due to the changes in the Os–C bond length. As already mentioned, the Os–C distances decrease with an increase of electron-withdrawing properties of the attached substituents. It can thus be expected that the shorter is the bond, the more covalent is its character, which implies an increase of |V(q)|magnitude.
However, this is compensated by an increase of G(q) which is related with Pauli repulsion between two closed shells. As a result, the H(q) varies within a very small range. These changes are relatively small because of a limited possibility of the influence of substituents on the Os–C bond, and can be more significant in the case of a larger spectrum of variability of a given bond.
Os–C(O) bond
The introduction of a given substituent into position 4 of the osmabenzene ring results in small but systematic changes in the Os–C (carbonyl) bonding. These values indicated, in the case of all Os–Ccarbonyl bonds ∇2ρ values at corresponding BCPs are positive, as it was found for closed-shell interactions.
On the other hand, the H(ρ) values are negative, as found for shared interactions. This is in agreement with observations made for the Ti–C bonds in titanium complexes [44], in the case when the metal–ligand bonding has a characteristic that represents a mix of the closed-shell and shared parameters.
Moreover, the H(ρ) values are more negative for Os–C5 bonds, which is directly connected with relative greater predominance of |V(q)| magnitude over the G(q) magnitude. This suggests a more covalent character of the Os–C5 bond as compared with the Os-C1, and is also in line with general knowledge, according to which low-field ligands (e.g. chlorine) weaken the cis placed M–C bonds. Generally, the greater value of |H(ρ)| (with negative sign), the more covalent character of the bond. It seems therefore that the covalent character of the Os –C(O) bonds increases with electron donating properties of the substituent attached to the osmabenzene.
This can be partially connected with the trans-effect and the fact that a relatively greater contribution of structure (I) in Figure 1 should lead to an increase of back-donation in the trans placed Os –Ccarbonyl bond. As a result, it can be said that in the analyzed organometallic species the Os –C(carbonyl) bonding has a more covalent character than the Os–C(osmabenzene) bonding.
Conclusion
The theoretical study of structure and properties in the osmabenzenes and substituted osmabenzenes indicated:
1. Singlet states are more stable than quintet states.
2. Ionization energy and electron affinity values increase in presence of withdrawing substituents.
3. The Os-C bond distances show that systematic variations in para substituted osmaabenzene complexes
4. Analysis of frontier orbitals shows the decreasing of the hardness and E(LUMO) in electron-withdrawing substituents.
5. There is a delocalized ϖ electron current on the basis of the NICS values. Also, aromaticity increases with electron-withdrawing substituents.
6. The QTAIM analysis indicates the covalent character of the Os-C5 bond is more than Os-C1. Also, this property of Os–C(O) bonds increases with electron donating properties of the substituent.
References
1. Bleeke, J. R. Chem. Rev 2001, 101, 1205-1227. [ Links ]
2. Wright, L. J. J. Chem. Soc. Dalton Trans. 2006, 1821-1827. [ Links ]
3. He, G.; Xia, H.; Jia, G. Chin. Sci. Bull 2004, 49, 1543-1553. [ Links ]
4. Jacob, V.; Landorf, C. W.; Zakharov, L. N.; Weakley, T. J. R.; Haley, M. M. Organometallics 2009, 28, 5183-5190. [ Links ]
5. Ghiasi, R.; Mokarram, E. E. Russian. J. Coord. Chem. 2011, 37, 463-467. [ Links ]
6. Ghiasi, R. Russian. J. Coord. Chem 2011, 37, 72-76. [ Links ]
7. Landorf, C. W.; Haley, M. l. M. Angew. Chem. Int. Ed. 2006, 45, 3914-3936. [ Links ]
8. Elliott, G. P.; Roper, W. R.; Waters, J. M. J. Chem. Soc., Chem. Commun. 1982, 811. [ Links ]
9. Iron, M. A.; Martin, J. M. L.; Boom, M. E. v. d. J. Am. Chem. Soc. 2003, 125, 13020-13021. [ Links ]
10. Gong, L.; Chen, Z.; Lin, Y. i.; He, X.; Wen, T. B.; Xu, X.; H.Xia: Chem. Eur. J. 2009, 15, 6258-6266. [ Links ]
11. Bleeke, J. R. Chem. Rev. 2001, 101, 1205-1227. [ Links ]
12. Guomei, H. E.; Haiping, X.; Guocheng, J. I. A. Chinese Science Bulletin 2004, 49, 1543-1553. [ Links ]
13. Karton, A.; Iron, M. A.; Boom, M. E. v. d.; Martin, J. M. L. J. Phys. Chem. A 2005, 109, 5454-5462. [ Links ]
14. Johns, P. M.; Roper, W. R.; Woodgate, S. D.; Wright, L. J. Organometallics 2010, 29, 5358-5365. [ Links ]
15. Frisch, M. J.; Trucks, G. W.; Schlegel, H. B.; Scuseria, G. E.; Robb, M. A.; Cheeseman, J. R.; Montgomery, J. A.; Jr.; Vreven, T.; Kudin, K. N.; Burant, J. C.; Millam, J. M.; Iyengar, S. S.; Tomasi, J.; Barone, V.; Mennucci, B.; Cossi, M.; Scalmani, G.; Rega, N.; Petersson, G. A.; Nakatsuji, H.; Hada, M.; Ehara, M.; Toyota, K.; Fukuda, R.; Hasegawa, J.; Ishida, M.; Nakajima, T.; Honda, Y.; Kitao, O.; Nakai, H.; Klene, M.; Li, X.; Knox, J. E.; Hratchian, H. P.; Cross, J. B.; Adamo, C.; Jaramillo, J.; Gomperts, R.; Stratmann, R. E.; Yazyev, O.; Austin, A. J.; Cammi, R.; Pomelli, C.; Ochterski, J. W.; Ayala, P. Y.; Morokuma, K.; Voth, G. A.; Salvador, P.; Dannenberg, J. J.; Zakrzewski, V. G.; Dapprich, S.; Daniels, A. D.; Strain, M. C.; Farkas, O.; Malick, D. K.; Rabuck, A. D.; Raghavachari, K.; Foresman, J. B.; Ortiz, J. V.; Cui, Q.; Baboul, A. G.; Clifford, S.; Cioslowski, J.; Stefanov, B. B.; Liu, G.; Liashenko, A.; Piskorz, P.; Komaromi, I.; Martin, R. L.; Fox, D. J.; Keith, T.; Al-Laham, M. A.; Peng, C. Y.; Nanayakkara, A.; Challacombe, M.; Gill, P. M. W.; Johnson, B.; Chen, W.; Wong, M. W.; Gonzalez, C.; Pople, J. A. Gaussian 03. Revision B.03 ed.; Gaussian, Inc., Pittsburgh PA, 2003. [ Links ]
16. McLean, A. D.; Chandler, G. S. J. Chem. Phys. 1980, 72, 5639-5648. [ Links ]
17. Krishnan, R.; Binkley, J. S.; Seeger, R.; Pople, J. A. J. Chem. Phys. 1980, 72, 650-654. [ Links ]
18. Hay, P. J.; Wadt, W. R. J. Chem. Phys 1985, 82, 299-310. [ Links ]
19. Hay, P. J.; Wadt, W. R. J. Chem. Phys 1985, 82 284-299. [ Links ]
20. Schaefer, A.; Horn, H.; Ahlrichs, R. J. Chem. Phys 1992 97, 2571-2577. [ Links ]
21. Hay, P. J.; Wadt, W. R. J. Chem. Phys 1985, 82 270-283. [ Links ]
22. Adamo, C.; Barone, V. J. Chem. Phys. 1998, 108, 664. [ Links ]
23. Schleyer, P. v. R.; Maerker, C.; Dransfeld, A.; H.Jiao; Hommes, N. J. R. v. E. J. Am. Chem. Soc. 1996, 118, 6317-6318. [ Links ]
24. Cyranski, M. K.; Krygowski, T. M.; Wisiorowski, M.; Hommes, N. J. R.; Schleyer, P. v. R. Angew. Chem., Int. Ed. 1988, 37, 177. [ Links ]
25. Bader, R. F. W. AIM2000 Program. ver 2.0, ed. Hamilton, Mc-Master University, 2000. [ Links ]
26. Pearson, R. G. Chemical Hardness; Wiley-VCH: Oxford, 1997. [ Links ]
27. Parr, R. G.; Yang, W. Density-Functional Theory of Atoms and Molecules; Oxford University Press: New York, 1989. [ Links ]
28. Parr, R. G.; L, v. S. l.; Liu, S. J. Am. Chem. Soc. 1999, 121, 1922. [ Links ]
29. Schleyer, P. v. R.; Maerker, C.; Dransfeld, A.; Jiao, H.; Hommes, N. J. R. v. E. J Am. Chem. Soc. 1996, 118, 6317. [ Links ]
30. Sobczyk, L.; Grabowski, S. J.; Krygowski, T. M. Chem. Rev. 2005, 105 3513. [ Links ]
31. Bader, R. F. W.; Matta, C. F.; Cortés-Guzman, F. Organometallics 2004, 23, 6253. [ Links ]
32. Fradera, X.; Austen, M. A.; Bader, R. F. W. J. Phys. Chem. A 1999, 103 304. [ Links ]
33. Bader, R. F. W.; Fang, D.-F. J. Chem. Theor. Comput. 2005, 1, 403. [ Links ]
34. Mitrasinovic, P. M. Can. J. Chem. 2003, 81, 542. [ Links ]
35. Pendas, A. M.; Blanco, M. A.; Francisco, E. Chem. Phys. Lett. 2006, 16, 417. [ Links ]
36. Poater, J.; Sola, M.; Bickelhaupt, F. M. Chem. Eur. J. 2006, 12, 2902. [ Links ]
37. Kovacs, A.; Esterhuysen, C.; Frenking, G. Chem. Eur. J. 2005, 11, 1813. [ Links ]
38. O'Brien, S. E.; Popelier, P. L. A. Can. J. Chem. 1999, 77, 28. [ Links ]
39. Howard, S. T.; Krygowski, T. M. Can. J. Chem. 1997, 75, 1174. [ Links ]
40. Bader, R. W. F.; Matta, C. F. Organometallics 2004, 23, 6253. [ Links ]
41. Melchor, V. S.; Alkorta, I.; Elguero, J.; Sundberg, M. R.; Dobado, J. A. Organometallics 2006, 25 5638. [ Links ]
42. Gonzalez, L.; Mo, O.; Yanez, M.; Elguero, J. J. Mol. Struct. 1996, 371, 1. [ Links ]
43. R.F.W. Bader: Atoms in Molecules: A Quantum Theory; Clarendon Press: Oxford, 1994. [ Links ]
44. Palusiak, M. J. Organometallic. Chem 2005, 692, 3866-3873. [ Links ]

