










Servicios Personalizados
Revista
Articulo
 Inglés (pdf)
Inglés (pdf)
 Artículo en XML
Artículo en XML Referencias del artículo
Referencias del artículo
 Enviar artículo por email
Enviar artículo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO -
 Accesos
Accesos
Links relacionados
-
 Similares en
SciELO
Similares en
SciELO
Compartir

 Permalink
PermalinkJournal of the Mexican Chemical Society
versión impresa ISSN 1870-249X
J. Mex. Chem. Soc vol.56 no.3 Ciudad de México jul./sep. 2012
Artículo
Nucleophilic Attack at the Pyridine Nitrogen Atom in a Bis(imino)pyridine System: The Local Hard and Soft Acids and Bases Principle Perspective
Arlette Richaud and Francisco Méndez*
Departamento de Química, División de Ciencias Básicas e Ingeniería, Universidad Autónoma Metropolitana-Iztapalapa, A.P. 55-534, México, D.F., 09340 México.
Received January 06, 2012.
Accepted June 04, 2012.
Abstract
Motivated by the unprecedented nucleophilic attack at the pyridine nitrogen atom in bis(imino)pyridines, observed by Gambarotta et al. and Gibson et al., and the fact that normally pyridine undergoes reactions with electrophiles at its nitrogen atom, we applied the local hard and soft acids and bases principle to assert that nucleophilic attack at pyridine nitrogen atom depends fundamentally on stereoelectronic factors.
Key words: Nucleophilic reactivity, bis(imino)Pyridines, Stereoelectronic Factors.
Resumen
Motivados por el ataque nucleofílico sin precedentes sobre el átomo de nitrógeno de la piridina en bis(imino)piridinas observado por Gambarotta et al. y Gibson et al. y por el hecho de que normalmente la piridina reacciona con electrófilos en el átomo de nitrógeno, aplicamos el principio de ácidos y bases duros y blandos para confirmar que el ataque nucleofílico al átomo de nitrógeno de la piridina depende fundamentalmente de factores estereoelectrónicos.
Palabras clave: Reactividad nucleofílica, bis(imino)piridinas, factores estereoelectrónicos.
1. Introduction
The diimine/pyridine ligand system enhances the reactivity of early and late transition metals toward olefin polymerization [1-3]. The six electron donor 2,6-bis(imino)pyridines 1 are tridentate ligands with potentially reactive nitrogen and carbon atoms (Figure 1) [4, 5].

In order to afford novel [N,N,N] ligands, Gibson et al. described the reaction of d6-bis(imino)pyridines 2 with MeLi which provides the new ligand 3 arising from nucleophilic attack at the pyridine atom (Scheme 1). The N-methylation was unequivocally confirmed by a single-crystal X-ray diffraction experiment [6]. In a parallel study, Gambarotta et al. found that reaction of bis(imino)pyridine with MeLi leads to the surprising alkylation at the pyridine nitrogen atom [7]. These reactions were unprecedented because pyridine derivatives in general undergo reactions with nucleophiles at the α and γ positions or, under special circumstances at the β positions of the pyridine ring.
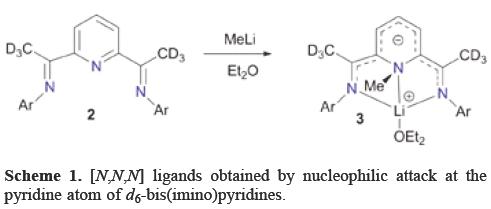
Gibson et al. surmised that "This highly selective nucleophilic attack at pyridine nitrogen is believed to arise from a combination of favourable circumstances: (i) di-ortho substitution of the aryl ring Ar that effectively shields the imino-carbon from nucleophilic attack, (ii) deuterium substitution at the ketimine position disfavouring proton (deuteron) abstraction and (iii) chelation of MeLi by the pyridine and imino nitrogen donors places the MeLi in a favorable position for methyl migration to the pyridine nitrogen".
The local HSAB principle, proposed and proved by minimization of the grand canonical potential by Gázquez and Méndez [8, 9], is a successful principle of reactivity based on density functional theory (DFT) local descriptors (Fukui functionf(r) and local softness s(r)) [10-12]. It establishes that although the softest atom in molecule A is in general the most reactive site, there may be other less soft sites, which may become the most reactive sites, regulated by the softness of the reacting molecule B [8, 9]. The local HSAB principle has been widely used in studies on multiple reaction sites and in strong and weak chemical interactions [13-19].
In this study we reveal, using the local HSAB principle and DFT local descriptors, that nucleophilic attack at the pyridine nitrogen atom of 1 depends fundamentally on stereoelectronic factors (for a description of stereolectronic effects see Juaristi and Cuevas [20].
Computational Details
The geometries of the minima of the potential energy surface for the reference molecules 4, 5a, and 6 were calculated at the B3LYP/6-311+G(2d,2p) level of theory using GAUSSIAN03 [21]. For 5b and 7, the experimental geometry obtained by X-ray diffraction [6] was used. The nucleophilic Fukui function f+(r) and local softness s+(r) for 4-7 were calculated using f+(r) = ρN+1(r) - ρN(r) and s+(r) = Sf+(r), where S is the global softness (inverse of the chemical hardness η) and ρN(r) and ρN+1(r) are the electronic density of the reference molecule and its corresponding anion respectively. The η value was obtained from the ionization potential (I) and the electron affinity (A), η = (I - A)/2. The f+(r) isosurface diagrams were plotted with GaussView [22].
Results and Discussion
In order to observe systematically how the pyridine nitrogen atom acquires electrophilic character and analyze the proposals made by Gibson et al. (i.e. i-iii), we studied the precursor 2,6-diacetyl-pyridine 4 (R=CH3), the [N,N,N] ligand bis(imino)-pyridine 5 {[2,6-(ArNCR)2C5H3N]} [R=CH3, Ar=Ph, 2,6-i-Pr2C6H3], the complex with MeLi {[2,6-(ArNCCH3)2C5H3N]}LiMe [Ar=2,6-i-Pr2C6H3] 6, and the complex with MeLi and Et2O {[2,6-(ArNCCH3)2C5H3N]}Li(Me)(Et2O) [Ar = 2,6-i-Pr2C6H3] 7 (Figure 2).
The analysis of Figure 3 shows the nucleophilic Fukui function f+(r) isosurface diagram for 4 (magenta colors correspond to positive f+(r) values; cutting value 0.01 a.u.). The diagram shows the reactive sites for 4 to a given soft nucleophile. According to the local HSAB principle, the softness of the nucleophile should be within the same range or value as the preferred site in order to react in the studied molecule, i.e. s+(r) = Sf+(r). Higher f+(r) values are located at the β positions of the ring followed by the carbonyl group of 4; no contribution is observed for the pyridine nitrogen atom. According to the local HSAB principle, the β-position and the carbonyl carbon atom will react with soft and less soft nucleophiles respectively. In a fashion analogous to the 1,4-Michael addition of nucleophiles to α,β-unsaturated electrophiles [13,18,19], as observed experimentally [6], the slightly soft nucleophile aniline (hardness η = 4.4 eV) [23] attacks the carbonyl carbon atom to yield 5a.

The conformational analysis of 5a shows that the s-trans,s-trans conformer 5at (Figure 4), is more stable by 12 kcal mol-1 than the s-cis,s-cis conformation 5ac (Figure 5) However, only in conformer 5ac, the pyridine nitrogen atom has a contribution to f+(r) (cf. Figures 4 and 5).


From Figure 5 it can be seen that the s-cis,s-cis-conformer 5ac has the highest f+(r) value at the pyridine nitrogen atom, followed by the imino nitrogen atoms. According to the local HSAB principle, the pyridine nitrogen atom will be attacked by the methyl anion, a soft nucleophile (η = 4.0 eV) [6, 7, 24].
Analyzing the calculated values of the f+(r) for 5b at the experimental geometry of 7 (s-cis,s-cis-conformation; Ar = 2,6-i-Pr2C6H3), we observe in Figure 6 the same behavior of the f+(r) values as in 5ac; the highest f+(r) values are located at the pyridine nitrogen atom.

Since 5ac has no ortho substitution in the N-phenyl ring, the nucleophilic attack at the pyridine nitrogen atom does not depend on steric factors. Furthermore, the electrophilic behavior of the pyridine nitrogen atom of 5 arises in the s-cis,s-cis-conformation and not in the s-trans,s-trans-geometry. Therefore, the nucleophilic attack at the pyridine nitrogen depends fundamentally on stereoelectronic rather than on steric factors (cf. i). The asseveration is corroborated in Figure 7. The f+(r) isosurface diagram for complex 6 shows that chelation of MeLi by the pyridine and imino nitrogen atoms does not modify the f+(r) behavior in the N,N,N ligand; the soft pyridine nitrogen atom continues to be the most reactive site in the molecule for the methyl nucleophilic attack. As a result, the chelation just places the MeLi in a favourable position for methyl migration to the pyridine nitrogen according to the proposal made by Gibson (cf. iii). Calculations at the B3LYP/6-311++G(2df,2pd)// B3LYP/3-21G level of theory showed that transfer of the Li-bound Me group to the nitrogen atom had low barrier of 5 kcal mol-1 and was exothermic by 26 kcalmol-1; the reverse reaction would have a barrier of 31 kcal mol-1 [7]. Gambarotta et al. also reported that pyridine ring becomes slightly more folded and receives more negative charge in the presence of a diethyl ether molecule coordinated to the lithium atom. The reported Mulliken charge on the ligand by the absence and presence of the ether molecule was -0.58 and -0.62 respectively.

Once the methyl group makes the nucleophilic addition to the pyridine nitrogen atom, the higher f+(r) values for 7 are located at the imino groups (Figure 8). Even while 7 is stable for extended periods at room temperature, the ligand was easily deprotonated at the imine methyl group with loss of methane [7, 25]. Gibson et al. [25] provided strong evidence that this transformation occurred via the re-formation of MeLi which dissociates from the ligand and is then available to dedeuterate the ligand backbone. Additionally, the methyl group could be abstracted from the nitrogen atom by the mild electrophiles i-PrBr and FeCl3, indicating that the alkylation at the nitrogen atom may be reversible.

Concluding remarks
The nucleophilic N-alkylation on pyridine is an unprecedented reaction throughout the history of chemistry, as Gibson, Gam-barotta, and coworkers have pointed out. The local HSAB principle and the DFT local descriptors suggest that nucleophilic attack at the pyridine nitrogen atom of 2,6-bis(imino)pyridines 1 depends fundamentally on stereoelectronic rather than on steric factors. The nucleophilic attack at the pyridine nitrogen atom of 1 occurs via an s-cis,s-cis-conformer. The ortho substitution in the aryl ring should not modify the nitrogen pyridine reactivity. The chelation of MeLi by the pyridine and imino nitrogen atoms does not modify the electrophilic behavior of the [N,N,N] ligand; the soft pyridine nitrogen atom continues to be the most reactive site in the molecule for the methyl anion nucleophilic attack.
Acknowledgments
We thank CONACyT-México for financial support (F.M.) and Dr. Eduardo González (UAM-I, México) for many helpful discussions. We thank referee (1) for the suggestions for improving the article.
References
1. Reardon, D.; Conan, F.; Gambarotta, S.; Yap, G.; Wang, Q. J. Am. Chem. Soc., 1999, 121, 9318-9325. [ Links ]
2. Gibson, V. C.; Kimberley, B. S.; White, A. P. J.; Williams, D. J.; Howard, P. J. Chem. Commun. 1998, 313-314. [ Links ]
3. Britovsek, G. J. P.; Gibson, V. C.; Kimberley, B. S.; Maddox, P. J.; McTavish, S. J.; Solan, G. A.; White, A. P. J.; Williams, D. J. Chem. Commun. 1998, 849-850. [ Links ]
4. Small, B. L.; Brookhart, M.; Bennett, M. A. J. Am. Chem. Soc., 1998, 120, 4049-4050. [ Links ]
5. Bruce, M.; Gibson, V. C.; Redshaw, C.; Solan, G. A.; White, A. J. P.; Williams, D. J.; Chem. Commun. 1998, 2523-2524. [ Links ]
6. Clentsmith, G. K. B.; Gibson, V. C.; Hitchcock, P. B.; Kimberley, B. S.; Rees, C. W. Chem. Commun. 2002, 1498-1499. [ Links ]
7. Khorobkov, I.; Gambarotta, S.; Yap, G. P. A.; Budzelaar, P. H. M. Organometallics 2002, 21, 3088-3090. [ Links ]
8. Gâzquez, J. L.; Méndez, F. J. Phys. Chem. 1994, 98, 4591-4593. [ Links ]
9. Méndez, F.; Gâzquez, J. L.; J. Am. Chem. Soc. 1994, 116, 9298-9301. [ Links ]
10. Ayers, P. W.; Parr, R. G. J. Am. Chem. Soc. 2001, 123, 2007-2017. [ Links ]
11. Chattaraj, P. K. J. Phys. Chem. A 2001, 105, 511-513. [ Links ]
12. Parr, R. G.; Yang, W. "Density Functional Theory of Atoms and Molecules", Oxford University Press, Oxford: UK, 1989. [ Links ]
13. Chandrakumar, K. R. S.; Pal, S. J. Phys. Chem. B 2001, 105, 4541-4544. [ Links ]
14. Méndez, F.; Galvàn, M.; Garritz, A.; Vela, A.; Gàzquez; J. L. J. Mol. Struct. (THEOCHEM), 1992, 211, 81-86. [ Links ]
15. Gregus, Z.; Roos, G.; Geerlings, P.; Nemeti, B. Toxicol. Sci., 2009, 110, 282-292. [ Links ]
16. Pal, S.; Chandrakumar, K. R. S. J. Am. Chem. Soc. 2000, 122, 4145-4153. [ Links ]
17. Geerlings, P.; De Proft. F.; Langenaeker, W. Chem. Rev. 2003, 103, 1793-1874. [ Links ]
18. Poon, T.; Mundy, B. P.; Shattuck, T. W. J. Chem. Ed. 2002, 79, 264-267. [ Links ]
19. Gilardoni, F.; Weber, J.; Chermete, H.; Ward, T. R.; J. Phys. Chem. 1998, 102, 3607-3613. [ Links ]
20. Juaristi, E.; Cuevas, G.; Acc. Chem Res. 2007, 40, 961-970. [ Links ]
21. Frisch, M. J.; Trucks, G. W.; Schlegel, H. B.; Scuseria, G. E.; Robb, M. A.; Cheeseman, J. R.; Montgomery, J. A. Jr.; Vreven, T.; Kudin, K. N.; Burant, J. C.; Millam, J. M.; Iyengar, S. S.; Tomasi, J.; Barone, V.; Mennucci, B.; Cossi, M.; Scalmani, G.; Rega, N.; Petersson, G. A.; Nakatsuji, H.; Hada, M.; Ehara, M.; Toyota, K.; Fukuda, R.; Hasegawa, J.; Ishida, M.; Nakajima, T.; Honda, Y.; Kitao, O.; Nakai, H.; Klene, M.; Li, X.; Knox, J. E.; Hratchian, H. P.; Cross, J. B.; Bakken, V.; Adamo, C.; Jaramillo, J.; Gomperts, R.; Stratmann, R. E.; Yazyev, O.; Austin, A. J.; Cammi, R.; Pomelli, C.; Ochterski, J. W.; Ayala, P. Y.; Morokuma, K.; Voth, G. A.; Salvador, P.; Dannenberg, J. J.; Zakrzewski, V. G.; Dapprich, S.; Daniels, A. D.; Strain, M. C.; Farkas, O.; Malick, D. K.; Rabuck, A. D.; Raghavachari, K.; Foresman, J. B.; Ortiz, J. V.; Cui, Q.; Baboul, A. G.; Clifford, S.; Cioslowski, J.; Stefanov, B. B. ; Liu, G.; Liashenko, A.; Piskorz, P.; Komaromi, I.; Martin, R. L.; Fox, D. J.; Keith, T.; Al-Laham, M. A.; Peng, C. Y.; Nanayakkara, A.; Challacombe, M.; Gill, P. M. W.; Johnson, B.; Chen, W.; Wong, M. W.; Gonzalez, C.; Pople, J. A. Gaussian 03, Revision C. 02; Gaussian, Inc.:Wallingford, CT, 2004. [ Links ]
22. GaussView 3.0, Gaussian, Inc. Carnegie Office Park - Building 6 Pittsburg PA 15106 USA, Copyright (c) 2000-2003 Semichem, Inc. [ Links ]
23. The trend of the experimental values for the halogen hardness, places the C6H5NH2 and Me nucleophiles as soft and softer groups respectively: ηF (7.01 eV) > ηCl (4.70 eV) > η C6H5NH2 (4.40 eV) > ηBr (4.24 eV) > ηMe (4.0 eV) > ηI (3.70 eV), see Pages 75, 80 and 86, Pearson, R. G. "ChemicalHardness", Wiley-VCH, Weinheim: Germany, 1997. [ Links ]
24. Parr, R. G.; Pearson, R.G. J. Am. Chem. Soc., 1983, 105, 7512-7516. [ Links ]
25. Blackmore, I. J.; Gibson, V. C.; Hitchcock, P. B.; Rees, C. W.; Kimberley, B. S.; Williams, D. J.; White, A. P. J. J. Am. Chem. Soc., 2005, 127, 6012-6020. [ Links ]
* The asterisk indicates the name of the author to whom inquires about the paper should be addressed.
Dedicated to José Luis Gázquez.

