










Services on Demand
Journal
Article
 English (pdf)
English (pdf)
 Article in xml format
Article in xml format Article references
Article references
 Send this article by e-mail
Send this article by e-mailIndicators
-
 Cited by SciELO
Cited by SciELO -
 Access statistics
Access statistics
Related links
-
 Similars in
SciELO
Similars in
SciELO
Share

 Permalink
PermalinkJournal of the Mexican Chemical Society
Print version ISSN 1870-249X
J. Mex. Chem. Soc vol.56 n.3 Ciudad de México Jul./Sep. 2012
Artículo
Auxiliary Density Perturbation Theory for Restricted Open-Shell Systems
Gregorio Guzmán-Ramírez,1 Francisco José Tenorio,2 Jaime Gustavo Rodríguez-Zavala,2 Roberto Flores-Moreno*1
1 Departamento de Química, Centro Universitario de Ciencias Exactas e Ingeniería, Universidad de Guadalajara, Blvd. Marcelino García Barragán 1421,C.P. 44430, Guadalajara, Jalisco, México, roberto.floresm@red.cucei.udg.mx
2 Departamento de Ciencias Exactas y Tecnologías, Centro Universitario de Los Lagos, Universidad de Guadalajara, Enrique Díaz de León 1144, C.P. 47460, Los Lagos, Jalisco, México.
Received December 06, 2011.
Accepted May 14, 2012.
Abstract
The recently developed approach to auxiliary density perturbation theory (J. Chem. Phys. 2008, 128, 134105) for the purpose of calculating molecular properties is here extended to include open-shell systems. Both unrestricted and restricted formalisms are considered. A linear equation system, twice as large as the auxiliary function set is obtained in both cases. For the first time, the formulation for auxiliary density perturbation theory for restricted open-shell formalism is derived.
Key words: Auxiliary Density, Perturbation, Theory, Open-shell.
Resumen
La teoría de perturbaciones de densidad auxiliar recientemente desarrollada (J. Chem. Phys. 2008, 128, 134105) para el cálculo de propiedades moleculares es extendida en este trabajo para el caso de sistemas decapa-abierta. Se consideran formalismos tanto restringidos como sin restricciones. En ambos casos se obtiene un sistema de ecuaciones lineales dos veces más grande que el conjunto de la función auxiliar. Por primera vez, se deriva la formulación de la teoría de perturbaciones de densidad auxiliar dentro del formalismo de capa-abierta restringido.
Palabras clave: Densidad auxiliar, perturbación, teoría, capa-abierta.
Introduction
The Kohn-Sham (KS) approximation [1] to density functional theory (DFT) [2, 3] is a very successful theoretical approach. Thousands of applications have been described for this methodology. Even though there are well known issues to be addressed concerning the essentials of the exchange-correlation functionals used [4-9], we believe that KS-DFT will enjoy popularity for a number of years more and thousands of new applications will be discovered. Therefore, rather than either applying or benchmarking DFT results or discussing its range of applications; the purpose of this paper is to provide an extra tool for calculating the molecular properties of the KS-DFT framework.
Very efficient implementations of KS-DFT can be achieved using the variational approximation of the Coulomb potential (VACP) [10-13]. As an example one can cite the Lowdin [14] and deMon2k programs [15]. Another approximation, closely related to the VACP, where auxiliary density is obtained by applying VACP is used for the direct calculation of the exchange-correlation energy and potential [16-19]. A well-defined energy expression and the corresponding gradients and higher order derivatives can be obtained from the resulting auxiliary density functional theory (ADFT). The accuracy of ADFT falls within the intrinsic accuracy of current KS-DFT implementations [18]. The computational efficiency of energy and structure optimization calculations when applying ADFT arises from the fact that the approximated density is a linear combination of auxiliary functions, and therefore the density calculation at each grid point is on a linear scale.
Analytic calculation of molecular properties, applying electronic structure methods is based on static linear response. The formulation of the so-called coupled perturbed Kohn-Sham equations [20-23] has been adapted from the analogous coupled-perturbed Hartree-Fock equations [24-27]. Another, density-based method eliciting linear response was thoroughly developed by McWeeny [28-33]. McWeeny's method is more amenable to DFT because it is more oriented towards density. In recent publications, a member of our group, together with Prof. Köster presented an explicit formulation for the evaluation of linear response dependent properties in ADFT. Derivation begins with McWeeny's formulation and includes the effects of ADFT approximations and auxiliary density. The resulting ADPT [34, 35] is very promising.
Due to the application of auxiliary density, self-consistency iterations are not required for ADPT. The efficiency of this method and its reliability has been tested for the calculation of polarizabilities [34, 35], Fukui functions [36, 37] and vibronic coupling [38]. Likewise, it was extended to manage time-dependent linear response [39]. Despite its success, ADPT formulation has not been completely documented. Working formulae for ADPT applied to closed-shell (ADPT-RKS) systems were described in Refs. [34-36, 39-41]. Extending this to cover the Pople-Nesbet [42, 43] unrestricted open-shell formulation (ADPT-UKS) was straightforward and the process was described in Ref. [35], although this is less accessible to the global scientific community. The least documented case occurs with reference to the restricted open-shell formulation (ADPT-ROKS), as this has never previously been documented.
In Ref. [35] it was reported that ADPT could not be combined with Roothaan's restricted open-shell method [44]. In this work, we finally indicate a method for implementing ADPT-ROKS, which is very important for applications to organic radicals, transition metal compounds and clusters. Furthermore, problems related to spin contamination may result in unreliable property calculations when applying ADPT-UKS. Although projection techniques [45, 46] could be used to cure spin contamination in SCF calculations, an adequate treatment of spin explicitly includes perturbations and therefore, the calculation of related properties requires a perturbation theory based on the restricted open-shell method [47-49]. Here we document ADPT-ROKS with the exclusive purpose of sharing it with those willing to take advantage of ADPT performance, in applications to restricted open-shell systems.
The manuscript is organized as follows. In Section 2, ADPT-ROKS formulation is outlined. Section 3 describes the extension to unrestricted open-shell systems for two purposes: firstly, to make its formulation widely available to the scientific community and secondly, to serve as a precedent for the derivation of ADPT-ROKS, which is described in detail in Section 4. Final remarks are presented in Section 5.
Results and Discussion
ADPT-RKS
For the sake of being comprehensive, the closed-shell case is also outlined here. Likewise, this serves as a starting point for introducing open-shell cases. A detailed description of ADFT can be found in Ref. [18]. Perturbation independent basis, auxiliary functions and local functionals are assumed for the sake of simplifying this presentation. However, even if these constraints were removed, we would still arrive at the same conclusions.
In the linear combination of Gaussian-type orbitals (LC-GTO) LCGTO ansatz the Kohn-Sham orbitals, Ψi (r) are expanded to become atomic orbitals, μ(r):

Because in a Kohn-Sham scheme the density, ρ(r) of a closed-shell system is presented as

Note the prefactor of 2, due to the double occupation of orbitals. Substitution of Eq. (1) into Eq. (2) yields:

where the closed-shell density matrix is defined as follows:
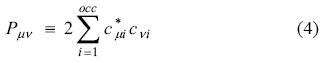
The auxiliary density used for the VACP,  (r), is built as a linear combination of the auxiliary functions k(r),
(r), is built as a linear combination of the auxiliary functions k(r),

The fitting coefficients xk are obtained by minimizing the density self-interaction error [10], resulting in the following linear equation system:

where the fitting coefficients are collected in the vector x, here we have introduced the Coulomb matrix G and the Coulomb vector J, whose elements are defined by [50]:

and
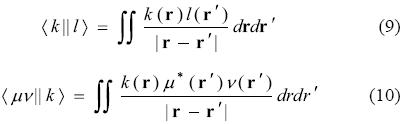
In this equation the symbol || stands for the coulomb operator 1/| r-r'| [18], therefore it is very important for readers to note that integral notations used here refer neither to those presented by Dirac, nor to those presented by Mulliken [51]. Note also that VACP is not exclusive to DFT calculations. Whenever a density is expressed as a bilinear form of the basis functions, an approximated density (expressed as a linear expansion of auxiliary functions) can be used to perform a VACP. This will be especially useful in the following sections.
The ADFT energy expression is [18]:
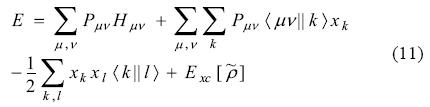
Hμv represents an element in the mono-electronic matrix. It includes kinetic energy, nuclear attraction integrals and any other external potential [18]. The Kohn-Sham matrix elements are obtained by differentiation of Eq. (11) with respect to the density matrix elements:
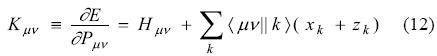
with the exchange-correlation fitting coefficients being defined

For the derivation of ADPT-RKS equations, we begin with the definition of the (first order) closed-shell perturbed density matrix as indicated in McWeeny's approach [32]:
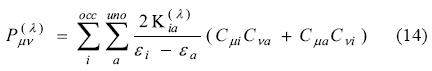
Here λ denotes the perturbation parameter, e.g. an electric field component in the calculation of polarizabilities, εi and εa refer to orbital energies of the ith occupied and ath unoccupied orbital.  represents the perturbed Kohn-Sham matrix in the reference molecular orbital representation,
represents the perturbed Kohn-Sham matrix in the reference molecular orbital representation,
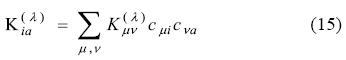
The derivative of the fitting equation system Eq. (6) with respect to the perturbation parameter λ, assuming perturbation independent basis and auxiliary functions, yields:

Substitution of Eq. (14) into Eq. (16) yields:
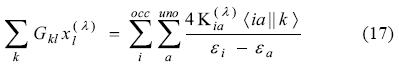
The perturbed Kohn-Sham matrix can be obtained in a straightforward way from the previously derived ADFT Kohn-Sham matrix Eq. (12):
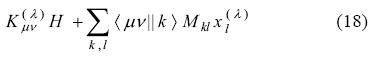
with

where f represents the exchange-correlation kernel in the auxiliary basis set representation, whose elements are
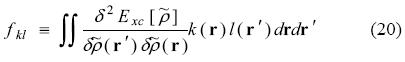
Substitution into Eq. (17) permits us to arrive an equation free of both P(λ) and K(λ),
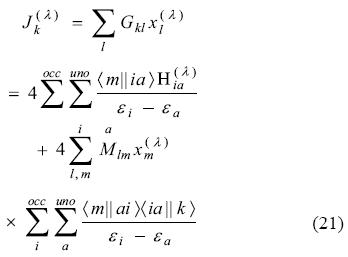
In order to simplify our notation, we now introduce the perturbation independent Coulomb coupling matrix A [34],

and the perturbation vector b(λ),
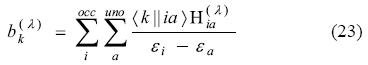
With these quantities and the Coulomb matrix defined in Eq. (22), equation (21) can be re-written as [34, 35]:

The perturbed fitting coefficients can then be obtained from this equation system. Since G, A and M are perturbation independent, only the perturbation vector b(λ) must be rebuilt for each different perturbation, e.g. the different electric field components in the case of a polarizability calculation. Eq. (24) was first derived in Refs. [34, 35]. Its dynamic counterpart has also been documented [35, 39]. Technical details and perturbation specific adjustments can be found in Refs. [34, 36, 38, 39].
ADPT-UKS
In the spin polarized ADFT the total density is given by the sum of the spin-densities. This fact can be expressed in terms of density matrices as follows:

Two independent Coulomb fitting procedures are performed for each spin-density. A total auxiliary density can be also constructed,

and each auxiliary spin-density is obtained from equations with the form

where s stands for spin a or ß. Likewise, for the Kohn-Sham matrices

with
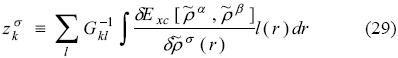
Applying McWeeny's theory, we get the response for each spin density matrix independently
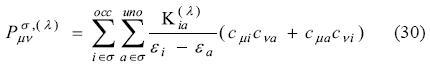
These relationships are obtained independently for each spin, due to the fact that each spin density is obtained from a separate Kohn-Sham matrix diagonalization. However, note that the factor of 2 before the Kohn-Sham matrix response has been removed, as there is only one electron with spin s for any given occupied orbital. Since the fitting is also independent for each spin, we also get two equation responses of the following type:

And, therefore, two relationships between the responses of the auxiliary density fitting coefficients and the corresponding Kohn-Sham matrix,
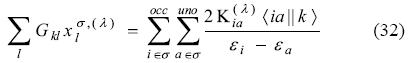
The coupling between alpha and beta sub-systems arises from the Coulomb and exchange-correlation contributions of the Kohn-Sham matrix response,

with

The exchange-correlation kernel is now spin-resolved,

Since 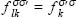 , there are 3 kernel matrices. Only
, there are 3 kernel matrices. Only  couples the α and β subsystems. The final equation system for ADPT-UKS is thus
couples the α and β subsystems. The final equation system for ADPT-UKS is thus

with the two Coulomb coupling matrices presented as
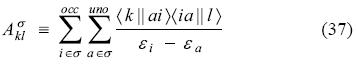
and the perturbation vector blocks

For a closed-shell system  and Eq. (36) reduces to Eq. (24). The following identification can be made for the closed-shell exchange-correlation kernel
and Eq. (36) reduces to Eq. (24). The following identification can be made for the closed-shell exchange-correlation kernel
This relation is relevant for the implementation of ADPT in ADFT codes, where the programming of exchange-correlation functionals is spin-resolved.
ADPT-ROKS
In a restricted open-shell Kohn-Sham formalism, a single effective Kohn-Sham matrix is diagonalized [33, 44, 52, 53]. The result of this diagonalization is a set of molecular orbitals which are doubly occupied and they are part of the closed-shell scheme. A second set, the open-shell, contains only single occupied orbitals. The third and last set includes all unoccupied (virtual) orbitals. The blocks of the effective Kohn-Sham matrix are built following this recipe [47, 54-56]:

where Kα and Kβ are the spin-resolved Kohn-Sham matrices built according to Eq. (28) and
The blocks of the effective Kohn-Sham matrix correspond to combinations of the three different molecular orbital blocks.
Consistent with McWeeny's theory, response to the Kohn-Sham matrix is required only for elements of the off-diagonal blocks. These are obtained as follows:
• Closed-open (i ∈ closed, n ∈ open):
• Closed-virtual (i ∈ closed, a ∈ virtual):

• Open-virtual (n ∈ open, a ∈ virtual):

Note that, the difference concerning the evaluation of the different types of KS matrix elements appear only on the exchange-correlation fitting coefficients. Substitution of Eq. (40) into Eq. (32) yields:
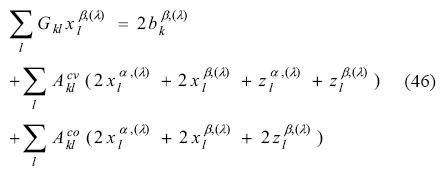
This time, the block coupling matrices are defined using super-scripts to identify the summation domains. They are presented as

As there are 3 off-diagonal blocks in the Kohn-Sham matrix response, three Coulomb coupling matrices emerged here. Differentiation of the exchange-correlation fitting coefficients leads us to the final ADPT-ROKS linear equation system
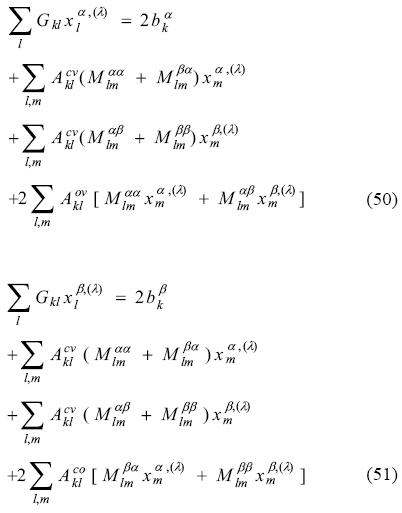
In matrix notation

Note that, in contrast to the UKS case, Eq. (52) is not symmetric with respect to spin. This originated from the coupling of the open-shell with the other two shells.
As the evaluation of A is the most demanding computational task in ADPT, the following relations are very useful for saving time and programming:

Since ROKS requires three different blocks, at least one of these should be explicitly calculated. The other two can be built using this one and the UKS blocks.
Conclusions
In this paper auxiliary density perturbation theory has been extended to include open-shell systems. For the first time ADPT equations for ROKS are presented. The derivation was presented showing a clear connection between ADPT-RKS and ADPT-UKS. A much more intimate relationship was discerned
between ADPT-UKS and ADPT-ROKS.
The characteristic feature of ADPT which makes possible a direct, non-iterative, solution was also found in the case of ADPT-UKS and ADPT-ROKS. However, for open-shell calculations, a system of equations twice the size of the auxiliary function set has to be solved.
This article contains all the documentation required for implementing ADPT for closed-shell, unrestricted open-shell and restricted open-shell cases. The only pre-requisite is the ADFT documentation [18].
The only limitation previously identified for ADPT, namely the application on restricted open-shell has been eliminated by following the approach described here. We are optimistic that this development will extend the range of applicability and impact of ADPT for the calculation of molecular properties.
Acknowledgments
RFM would like to acknowledge a number of enlightening discussions with Prof. Dr. Andreas M. Köster. This work was financially supported by CONACyT (Retención 2009: 117045 and CB 2009: 127362) and PROMEP 2011. GGR gratefully acknowledges a postdoc fellowship from CONACyT (Proyecto 138455).We would like also to acknowledge here the long standing effort of Prof. José Luis Gázquez for development of DFT research in México.
References
1. Kohn, W.; Sham, L. J. Phys. Rev. 1965, 140, A1133-A1138. [ Links ]
2. Hohenberg, P.; Kohn, W. Phys. Rev. 1964 136, B864-B871. [ Links ]
3. Parr, R. G.; Yang, W. Density Functional Theory of Atoms and Molecules Oxford University Press: New York, 1989. [ Links ]
4. Vela, A.; Medel V.; Trickey, S. B. J. Chem. Phys. 2009, 130, 244103-244108. [ Links ]
5. Proynov, E.; Vela, A.; Salahub, D.R. Chem. Phys. Lett. 1994, 230, 419-428. [ Links ]
6. Gázquez, J. L.; Cedillo, A.; Vela, A. J. Phys. Chem. A. 2007, 111, 1966-1970. [ Links ]
7. Gázquez, J. L. J. Mex. Chem. Soc. 2008, 52, 3-10. [ Links ]
8. Zhao, Y.; Truhlar, D. G. Theor. Chem. Acc. 2008, 120, 215-241. [ Links ]
9. Becke, A. D.; Arabi, A. A.; Kannemann, F. O. Can. J. Chem. 2010, 88, 1057-1062. [ Links ]
10. Dunlap, B. I. ; Connolly, J. W. D.; Sabin, J. R. J. Chem.Phys.(1979), 71, 3396/1-6. [ Links ]
11. Mintmire, J. W.; Dunlap, B. I. Phys. Rev. A. 1982, 25, 88-95. [ Links ]
12. Mintmire, J. W.; Sabin, J. R.; Trickey, S. B. Phys. Rev. B. 1982, 26, 1743 -1753. [ Links ]
13. Dunlap, B. I. J. Mol. Struct. (THEOCHEM) 2000, 529, 37-40. [ Links ]
14. Flores-Moreno, R.; González, S. A.; Aguirre, N. F.; Posada, E. F.; Romero, J.; Moncada, F.; Merino, G.; Reyes, A.Lowdin: A general code for the treatment of any quantum particle. 2011. [ Links ]
15. Köster, A. M.; Geudtner, G.; Calaminici, P.; Casida, M. E.; Domínguez, V. D.; Flores-Moreno, R.; Gamboa, G. U.; Goursot, A.; Heine, T.; Ipatov, A.; Janetzko, F.; del Campo, J. M.; Reveles, J. U.; Vela, A.; Zúñiga-Gutiérrez, B.; Salahub, D. R. deMon2k-Version 3, The demon developers, 2011 (Cinvestav, México City). http://www.demon-software.com [ Links ]
16. Laikov, D. N. Chem. Phys. Lett.1997, 281, 151-156. [ Links ]
17. Köster, A. M. Habilitation thesis, 1998, (Universität Hannover). [ Links ]
18. Köster, A. M.; Reveles, J. U.; del Campo, J. M. J. Chem. Phys. 2004, 121, 3417/1-7. [ Links ]
19. Birkenheuer, U. ;Gordienko, A.B.; Nasluzov, V. A.;Fuchs-Rohr, M. K.;Rösch, N lnt. J. Quantum Chem.2005 102, 743-761. [ Links ]
20. Founder, R. J. Chem. Phys. 1990, 92, 5422/1-8. [ Links ]
21. Komornicki, A.; Fitzgerald, G. J. Chem. Phys.1993, 98, 1398/1-24. [ Links ]
22. Handy, N. C.; Colwell, S. M.; Murray, C. W.; Amos, R. D. Chem. Phys. Lett. 1993, 210, 261-268. [ Links ]
23. Lee, A. M.; Colwell, S. M. J. Chem. Phys.1994, 101, 9704/1-6. [ Links ]
24. Gerratt, J. ; Mills, I. M.J. Chem. Phys. 1968, 49, 1719/1-11. [ Links ]
25. Schlegel, H. B.; Pople, J. A.; Krishnan, R.; Binkley, J. S. Int. J. Quantum Chem. Symp. 1979, 13, 225-241. [ Links ]
26. Karna, P.; Dupuis, M. J. Comput. Chem.1991, 12, 487-504. [ Links ]
27. Goddard, J. D.; Yamaguchi, Y.; Osamura, Y.; Schaefer III, H. F. A New Dimension to Quantum Chemistry: Analytic Dericative Methods in Ab Initio Molecular Electronic Structure Theory (Oxford University Press, New York, 1994. [ Links ]
28. McWeeny, R. Phys. Rev.1962, 126, 1028-1034. [ Links ]
29. Diercksen, G.;McWeeny, R. J. Chem. Phys.1966, 44, 3554/1-7. [ Links ]
30. McWeeny, R.;Diercksen, G. J. Chem. Phys.1968, 49, 4852/1-5. [ Links ]
31. Dodds, J. L.;McWeeny, R.;Raynes, W. T.; Riley, J. P. Mol. Phys. 1977, 33, 611-617. [ Links ]
32. Dodds, J. L.; McWeeny, R.; Sadlej, A. J. Mol. Phys. 1977, 34, 1779-1791. [ Links ]
33. McWeeny, R. Methods of Molecular Quantum Mechanics, Second reprinting Academic Press, London, 2001. [ Links ]
34. Flores-Moreno, R.; Köster, A. M. J. Chem. Phys.2008, 128, 134105/1-10. [ Links ]
35. Flores-Moreno, R. Ph.D.Thesis, 2006 (CINVESTAV, México). [ Links ]
36. Flores-Moreno, R. ;Melin, J.; Ortiz, J. V.; Merino, G. J. Chem. Phys.2008, 129, 224105/1-6. [ Links ]
37. Flores-Moreno, R. J. Chem. Theory Comput. 2010, 6, 48-54. [ Links ]
38. R. Flores-Moreno, E. Orozco-Guareño, 2011, Unpublished. [ Links ]
39. Carmona-Espíndola, J.; Flores-Moreno, R.;Köster, A. M. J. Chem. Phys.2010, 133, 084102/1-10. [ Links ]
40. Janetzko, F. ;Goursot, A.;Mineva, T.;Calaminici, P.; Flores-Moreno, R.;Köster, A. M.;Salahub, D. R.Nanoclusters: A Bridge Across Disciplines, Ed. P. Jena, A. Castleman, Jr., Elsevier, Amsterdam, 2010. [ Links ]
41. Calaminici, P.; Domínguez-Soria, V. D.; Flores-Moreno, R.; Gamboa-Martínez, G. U.; Geudtner, G.; Goursot, A.; Salahub, D. R.; Köster, A. M. Vademecum of Computational Chemistry 2011 (In press). [ Links ]
42. Pople, J. A.;Nesbet, R. K.J. Chem. Phys. 1954, 22, 571/1-2. [ Links ]
43. Binkley, J. S.;Pople, J. A.;Dobosh, P. A. Mol. Phys.1974, 28, 1423-1429. [ Links ]
44. Roothaan, C. C. J. Rev. Mod. Phys.1960, 32, 179-185. [ Links ]
45. Bacon, A. D.; M Zerner, C.Theor. Chim.Acta.1979, 53, 21-25. [ Links ]
46. Amos, T. ; Snyder, L. C. J. Chem. Phys. 1964, 41, 1773/1-11. [ Links ]
47. Rinkevicius, Z. ;Tunell, I.; Sa ek, P.;Vahtras, O.;Ågren, H. J. Chem. Phys.2003, 119, 34/1-13. [ Links ]
48. Rinkevicius, Z.;Telyatnyk, L.; Sa ek, P.;Vahtras, O.;Ågren, H. J. Chem. Phys. 2003, 119, 10489/1-8. [ Links ]
49. Rinkevicius, Z.; de Almeida, K. J.; Vahtras, O. J. Chem. Phys. 2008, 129, 064109/1-17. [ Links ]
50. Köster, A.M.;Calaminici, P.; Gómez, Z.; Reveles, U. in Reviews of Modern Quantum Chemistry, A Celebration of the Contribution of Robert G. Parr, Editor: K. Sen. World Scientific Publishing Co., New Jersey, 2002. [ Links ]
51. Szabo, A.; Ostlund, N. S. Modern Quantum Chemistry, Dover Publication Inc., Mineola New York, 1996. [ Links ]
52. Filatov, M.; Shaik, S. Chem. Phys. Lett. 1998, 288, 689-697. [ Links ]
53. Okazaki, I.; Sato, F.; Yoshihiro, T.; Ueno, T.; Kashiwagi, H. J. Mol. Struct. (THEOCHEM) 1998, 451, 109-119. [ Links ]
54. Hamilton, T. P.; Pulay, P. J. Chem. Phys.1986, 84, 5728/1-7. [ Links ]
55. Murray, C.; Davidson, E. R. Chem. Phys. Lett.1991, 187, 451-454. [ Links ]
56. Russo, T. V.; Martin, R. L.; Hay, P. J. J. Chem. Phys.1994, 101, 7729/1-9. [ Links ]
* The asterisk indicates the name of the author to whom inquires about the paper should be addressed.

