










Servicios Personalizados
Revista
Articulo
 Inglés (pdf)
Inglés (pdf)
 Artículo en XML
Artículo en XML Referencias del artículo
Referencias del artículo
 Enviar artículo por email
Enviar artículo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO -
 Accesos
Accesos
Links relacionados
-
 Similares en
SciELO
Similares en
SciELO
Compartir

 Permalink
PermalinkJournal of the Mexican Chemical Society
versión impresa ISSN 1870-249X
J. Mex. Chem. Soc vol.56 no.3 Ciudad de México jul./sep. 2012
Artículo
Theoretical quantum study about the adsorption of BH4- onto X(100) where (X = Cu, Ag and Au)
Luis Humberto Mendoza-Huizar,1* Diana Elizabeth García Rodríguez,1,2 Clara Hilda Ríos-Reyes,3 Alejandro Alatorre-Ordaz2
1 Universidad Autónoma del Estado de Hidalgo. Área Académica de Química. Ciudad del Conocimiento. Carretera Pachuca-Tulancingo Km. 4.5 Mineral de la Reforma, Hidalgo. C.P. 42186. hhuizar@uaeh.edu.mx
2 Universidad de Guanajuato. Departamento de Química. Cerro de la Venada S/N; Guanajuato, Gto. C.P. 36040.
3 Universidad Autónoma del Estado de Hidalgo. Área Académica de la Tierra y Ciencias de los Materiales. Ciudad del Conocimiento. Carretera Pachuca-Tulancingo Km. 4.5 Mineral de la Reforma, Hidalgo. C.P. 42186.
Received January 20, 2012.
Accepted April 11, 2012.
Abstract
In present work we analyzed some electronic properties involved during the adsorption of BH4- on Cu(100), Ag(100) and Au(100) surfaces. Reactivity descriptors such as ionization energy, hardness, electrophilicity, frontier molecular orbitals, condensed Fukui function, adsorption energies and density of states were calculated to identify changes in the reactivity on Cu(100), Ag(100) and Au(100). The results suggest the BH4- adsorption is favored on Cu(100) more than on Ag(100) or Au(100). The BH4- -Au(100) system showed higher values of μ and ω in comparison with BH--Ag(100) and BH4- Cu(100) systems. Last results suggest that gold is a better electron acceptor in comparison with silver and copper. Also, the fraction of electrons transferred during the BH4- adsorption was calculated indicating a bigger electron transfer from BH4- to Cu(100) compared to Au and Ag.
Key words: Borohydride, Adsorption, Cu, Ag, Au, DFT, LANL2DZ, DOS.
Resumen
En el presente trabajo analizamos algunas propiedades electrónicas involucradas durante la adsorción del BH4- sobre las superficies Cu(100), Ag(100) y Au(100). Se calcularon descriptores de reactividad tales como energía de ionización, dureza, electrofilicidad, orbitales moleculares frontera, función Fukui condensada, energías de adsorción y densidad de estados, para identificar cambios en la reactividad de las superficies Cu(100), Ag(100) y Au(100). Los resultados sugieren que la adsorción del BH- es favorecida sobre Cu(100) más que sobre Ag(100) y Au(100). El sistema BH4- -Au(100) mostró los valores mayores de μ y ω en comparación con los sistemas BH4- -Ag(100) y BH4- -Cu(100). Estos resultados sugieren que el oro es un mejor aceptor de electrones que plata o cobre. También se calculó la fracción de electrones durante el proceso de adsorción, los resultados indicaron que existe una mayor transferencia de carga del BH- hacia el cobre comparada con las obtenidas en Au y Ag.
Palabras Clave: Borohidruro, adsorción, Cu, Ag, Au, TFD, LANL-2DZ, DOS.
Introduction
A fuel cell (FC) is an attractive energy device which may operate continuously, as long as external fuel and oxidant reactants are supplied [1]. If this external fuel may be regenerated and coupled with a clean source of renewable energy; a FC represents a sustainable alternative to conventional energy sources [2]. Several FCs technologies have been developed like a proton exchange membrane FC which uses hydrogen to produce clean energy, water and heat. Other acidic FCs use methanol, ethanol, formic acid, hydrazine and glucose as fuels and their power generation depends on several factors such as catalyst, fuel purity, membrane material and cell design [3, 4].
The direct borohydride fuel cells (DBFC) have attracted a great interest because of their potential advantages such as a high capacity (5.7 Ah g-1) and energy density (9.3 Wh g-1 at 1.64 V) [5]. In these cells the borohydride or tetrahydroborate anions (BH4-) can be oxidized directly on a large variety of electrode materials liberating a maximum of eight electrons via the following suggested overall reaction [6]:
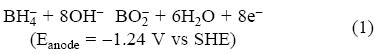
However, the final product of the surface oxidation reaction (1) still is unclear [7]. Different electrodes have been employed to carry out the borohydride oxidation but only on gold [7-10] and silver [7, 10, 11], it is possible to produce eight electrons [7, 11]. Although the borohydride oxidation on silver occurs at more positive potentials than on gold, silver is more attractive due to its lower cost (less than 2% compared to the gold's cost). Studies carried out by Chatenet et al. [7] showed transference of 7.5 electrons during the borohydride oxidation on polycrystalline silver employing a solution containing 1 M NaOH. Similar results have been reported employing oxidized polycrystalline silver [12], and chemical silver deposited on different carbon substrates [13]. From such studies, it was found the borohydride oxidation reaction (BOR) starts following an adsorption step (equation 2) and an electrochemical reaction (equation 3) [6]:
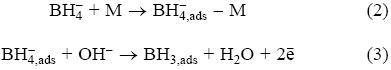
However, an understanding of the mechanisms involved in equations (2) and (3) is still missing. Thus, an analysis at electronic level would allow an efficient design of new materials to carry out the borohydride oxidation. From equation (2) note the first step previous to the oxidation reaction of BH4- is the borohydride adsorption, which may change the electronic properties of the substrate and its behavior during the oxidation route. Recently, the borohydride adsorption on Pt(111) and Au(111) surfaces was studied employing Density Functional Theory (DFT). It was found the initial adsorption step dictates the activity of gold and platinum anodes [8]. However, in such studies an analysis of the molecular reactivity involved during the adsorption was not done.
Seemingly silver and gold are the best electrodes to get a transfer of eight electrons during BOR. Here it is interesting to note that copper shows a similar electronic configuration to the presented by gold and silver. However, copper surface has not been employed as DBFC anodes. This is because of the thermodynamical oxidation potential of Cu (+0.159 V) is more positive than the BOR (-0.45 V). However, it is important to consider that an adsorption process may change the electronic properties of the surface including its oxidation potential. It has been reported that Cu, Ag and Au show a periodic pinpoint distribution of the electrophilic actives sites [15-17]. Thus, it is possible to suggest the BH4- adsorption on to these metals may show a periodic behavior. Therefore, in this work we examine some reactivity descriptors and the distribution of active sites of the monocrystalline surfaces of Cu, Ag and Au with (100) orientation caused by borohydride adsorption. We consider the results gained will allow getting a major understanding to select the substrates used during the BOR.
Theory
From DFT it is possible to define universal concepts of molecular stability and reactivity such as electronic chemical potential (μ), absolute hardness (η), and global electrophilicity index (ω) [18-24]. The electronic chemical potential μ was defined by Parr et al [25] as
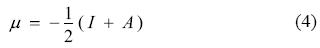
where, μ is characteristic of electronegativity of molecules, I is the vertical ionization energy and A stands for the vertical electron affinity. Absolute hardness can be defined as [26-28]:

Also, the global electrophilicity index ω was introduced by Parr [22, 29, 30,] and calculated using the electronic chemical potential μ and the chemical hardness η:

According to this definition ω measures the propensity of specie to accept electrons. Thus, a good nucleophile has low values of μ and ω. On the other hand, a good electrophile is characterized by high values of μ and ω. Also, the hard and soft acids and bases principle (HSAB) has been useful to predict the reactivity of chemical systems [27, 28, 30, 31-35]. The HSAB principle has been used in a local sense in terms of DFT concepts such as Fukui function f( ) [36, 37]. Gazquez and Mendez showed that sites in chemical species with the largest values of Fukui Function (f() ) are those with higher reactivity [36, 37]. The Fukui function is defined as [38-41]:
) [36, 37]. Gazquez and Mendez showed that sites in chemical species with the largest values of Fukui Function (f() ) are those with higher reactivity [36, 37]. The Fukui function is defined as [38-41]:
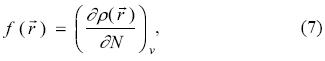
where ρ() is the electronic density, N is the number of electrons and v is the external potential exerted by the nucleus. This parameter is associated to the frontier orbital within the frozen core approximation. This approximation considers that when there is a variation on the number of electrons, only the respective frontier orbital is affected, when N increases to N + dN it is obtained the Fukui function for an electrophilic attack:
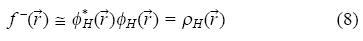
where ρH() is the electronic density of the highest occupied molecular orbital (HOMO).
When N diminish to N-dN it is obtained the Fukui function for an nucleophilic attack:
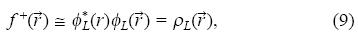
where ρL() is the electronic density of the lowest occupied molecular orbital (LUMO). The Fukui function is a local reactivity descriptor that indicates the preferred regions where a chemical species will change its density when the number of electrons is modified. It is possible to define the corresponding condensed or atomic Fukui functions on the jth atom site as [42-47]:
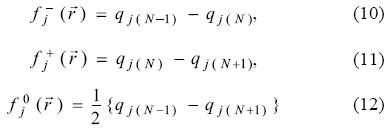
for an electrophilic  , nucleophilic
, nucleophilic  or free radical attack
or free radical attack  on the reference molecule, respectively. In these equations, qj is the atomic charge (evaluated from Mulliken population, electrostatic derived charge, etc.) at the jth atomic site in the neutral (N), anionic (N + 1) or cationic (N - 1) chemical species.
on the reference molecule, respectively. In these equations, qj is the atomic charge (evaluated from Mulliken population, electrostatic derived charge, etc.) at the jth atomic site in the neutral (N), anionic (N + 1) or cationic (N - 1) chemical species.
Results and discussion
Optimized geometry and reactivity descriptors
Figure 1 shows the optimized structures at B3LYP/LANL2DZ level at the adsorption site. It may be observed the bond lengths of BH4-- with Cu, Ag and Au increase respectively.
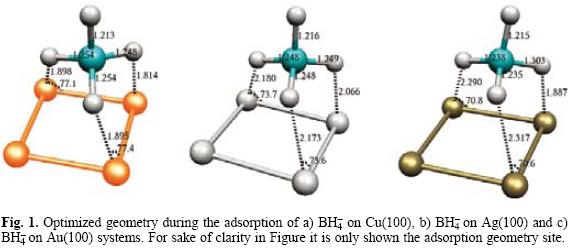
In all cases the H-B length was 1.25 A in average which compare favorably with the value of 1.24 A reported recently by Arevalo [48] during the adsorption of BH4-- on Mn and 1.24 A reported by Rostamikia et al for the the adsorption of BH4- on Au(111) and Pt(111) [49-51]. Also, the adsorption energy calculated for each system is reported in Table 1. The adsorption values reported are in the range reported by Arevalo [48] and Rostamikia et al [49-51] for the BH- on different surfaces. Note the system BH4--Cu(100) has the lowest adsorption energy compared to Ag and Au which suggests a favored adsorption of BH4- on copper surface. From same table, note the gap energy for BH4--Au(100) system is bigger in comparison with silver and copper suggesting more stability. If one compares the band gap values with the exhibited by the clean surfaces, it is possible to observe the BH4- adsorption stabilizes the BH4--X(100) systems.
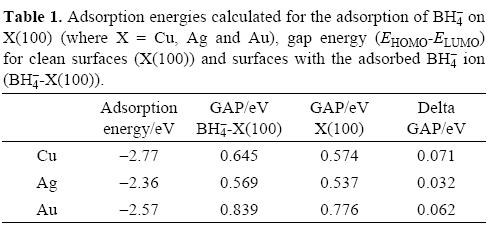
Reactivity descriptors such as μ, η and ω involved during the adsorption of BH4- onto X(100) were calculated and they are reported in Table 2. In all cases, observe the hardness value of the surfaces is decreased when the BH4- is adsorbed. This result indicates the adsorption activates the surfaces. Also, employing the values of η and μ one can calculate the global electrophilicity index ω given by equation (6) where this reactivity index measures the susceptibility of species to accept electrons. Observe that gold is being able to accept more electrons than either copper or silver. Additionally, the BH4--Au(100) system has the higher values of μ. and ω. Last result suggests that gold is more electronegative and a better electron acceptor when the BH4- ion is adsorbed. If one compares last results with the reactivity descriptors calculated for clean surfaces (Table 3), it is possible to note that when BH4- is adsorbed their capacity to accept electrons is diminished. On the other hand, the hardness of the systems decreased with the BH4- adsorption, which suggests an increase in the reactivity of the surfaces, however the change in the copper surface reactivity is few. It is seen that the BH4- adsorbed increases the ability of the surfaces to accept electrons (see ω in Table 2) and their reactivity, except for copper. From the parameters reported in Table 2, it is possible to calculate the electronegativity value (χ = ½(I + A) [18]. Also, it is possible to calculate the fraction of electrons transferred during the borohydride adsorption following the next equation [52].
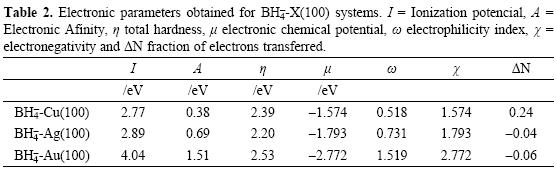
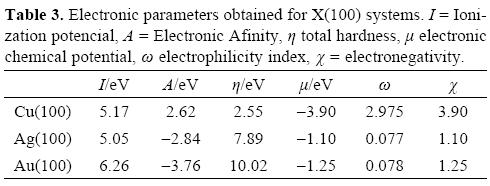
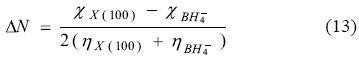
From the results reported in Table 2, note that ΔN is bigger onto copper in comparison with silver and gold which suggests a major electron transfer to this substrate.
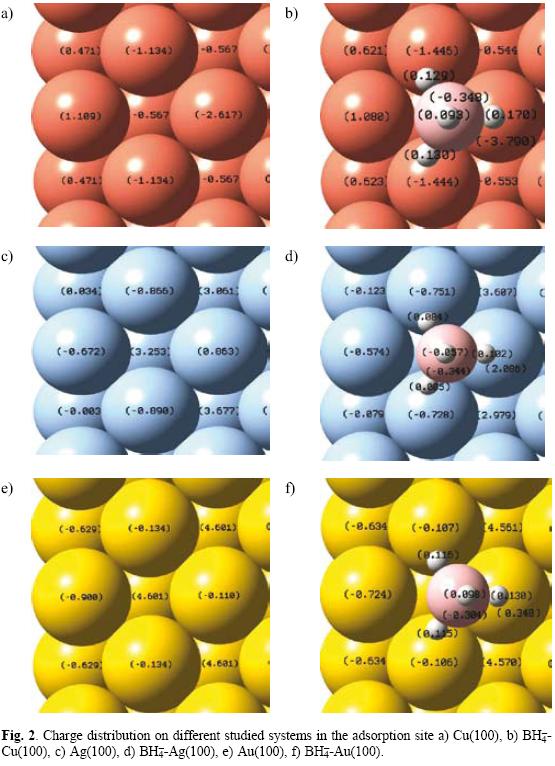
Charge distribution
It is well known the interaction among molecular species modifies the electronic density of such species causing a redistribution of the electric charges on them. It is because the magnitude of such redistribution is a function of the interaction. In the case of the adsorption, the change on the charge values will be indicative of such interaction. Figure 2 shows the charge distribution on Cu(100), Ag(100) and Au(100) surfaces and those where the BH4- is adsorbed. It is important to mention that when a cluster model is employed to study a surface, there is charge redistribution due to the border effects. Thus, there are two possible ways to take into account the border effects into the calculations. The first one is to saturate the valences on the cluster borders considering in artificial way the effect of the others atoms in the limits of the system. A second one consists in increasing the cluster size until the electronic properties do not change in the center of the cluster. For Cu(100) the variation in the charge values in the center of the cluster is less to 3% when the cluster size is bigger than four unit cells [16, 17]. Similar results have been obtained for gold and silver. Therefore, the surfaces modeled with clusters containing four cells should consider the border effects. Note that for BH4--Cu(100) (Figure 3b) there is, in the center of the cluster, an increase in the negative charge on the cluster compared with the clean surface (Figure 2a). Thus, in Cu(100) the atom with a charge of -2.617 got a value of -3.790 when the BH- was adsorbed indicating a charge transfer of 1.173 from BH4- to Cu(100). Note that a decreasing of the charge on BH4- is similar to an oxidation process caused by the adsorption. In the case of silver and gold an increase in the positive charge on the surface could be observed around the adsorption site. This is because there is a charge transfer from surface to BH4- which may be interpreted as a BH4- reduction process. Also, note that the charge transferred from gold to BH4- is major in comparison with the charge transferred from Ag to BH4-. Las results suggest that when the BH4- oxidation is performed it is possible to get more electrons from gold and silver in comparison with copper. Thus, it is a possible explanation why gold is more efficient to getting electrons during the borohydride oxidation.
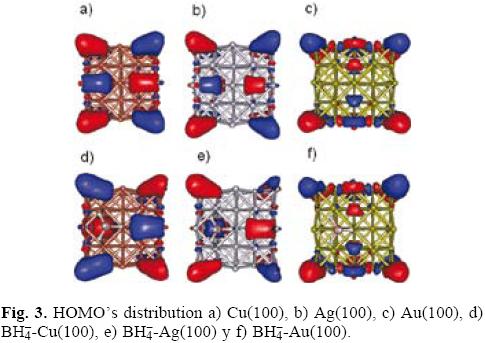
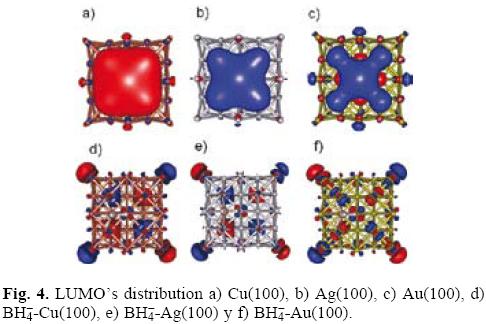
Molecular orbitals analysis
The distribution of the electrophilic sites in a system can be derived from the theory of frontier orbital within the frozen core approximation [31], Eq. (8). To find out this distribution caused by the borohydride adsorption, we analyzed the sites where the HOMO frontier orbital attains its larger absolute value on the studied surfaces. The greatest extension value of HOMO, calculated at B3LYP/LANL2DZ level, was observed on hollow positions; see Figure 3a, 3b and 3c for copper, silver and gold respectively. These results are consistent with the previously reported in literature [15-17]. Note the BH4- interaction with gold surface does not modify the HOMO's distribution and the number of electrophilic actives sites (NES) present in gold remains constant during the interaction. Different results were observed on copper and silver. On these surfaces if one considers the BH4- adsorption the number of NES decreases. This result indicates the electrophilic tendency attack is lesser on copper and silver when the BH4- ion is adsorbed. Last results are agreeing with the index ω value calculated in this work. In the case of LUMO's distribution for clean surfaces of copper (Figure 4a) and silver (Figure 4b), it was observed a clear delo-calization. Also, observe that this delocalization is bigger to the presented by gold surface (Figure 4c). Last behavior suggests that copper and silver are more reactive to a nucleophilic attack in comparison to gold. The modification of LUMO's distribution caused by the BH4- adsorption is depicted in Figure 4d, 4e and 4f for copper, silver and gold respectively. Note that BH4- deactivates the copper and silver surface. On the other hand, the gold surface is activated around the adsorption site.
Condensed Fukui function
From Figure 3 and 4 it may be observed that the HOMO and LUMO orbitals do not allow an analysis of the reactivity atom by atom. However, reactivity indexes derived from DFT theory have been successfully applied in describing and understanding of chemical reactivity defining atomic reactivity indexes, such as the condensed Fukui function. Thus, it possible to calculate the pin point distribution of the reactivity. We employed the equations (10-12) to calculate the value of the Fukui function atom by atom for electrophilic, nucleophilic and free radical attack. The values were coded as is shown in Table 4, the darker atoms corresponds to higher condensed Fukui function while the clearer atoms are associated to the lower values. For the electrophilic case, observe that the BH4- adsorption increases the reactivity on gold and copper, while deactivates the silver reactivity. For nucleophilic case the copper surface was the most activated compared with silver and gold. Also, we show the behavior of the surfaces for a radical free attack, note that copper and gold showed the higher reactivity.

Density of states
The density of states (DOS) provides a convenient overall view of the cluster electronic-structure. We studied the dependence on electronic properties of clusters by examining the calculated total electronic DOS. The total DOS of a cluster is composed by compact d states and the more expanded sp states. In small clusters the d and sp bands are clearly separated while the change of d states from small clusters toward bulk solid behavior is a monotonic broaden of band width. As the cluster size increases, both d and sp levels gradually broaden, shift, overlap with each other, and finally come into being bulk electronic band. At a first glance note the presence of continuous electronic bands, see Figure 5, which suggests that our models reproduce adequately the solid behavior. Also, from Figure 5, observe that in all cases the borohydride adsorption displaces the states occupation to higher energy levels in BH4-X(100) system in comparison with X(100). . Last may be indicative of a bigger stability caused by the adsorption. Note the LUMO-HOMO gaps are slightly enlarged, suggesting a major stability during the adsorption. The values of DOS at HOMO and LUMO energies almost were not modified. It has been reported that for a metal at absolute zero, the reciprocal of the hardness is the density of states at the Fermi level (g(εF)) [53-56], equation (14):
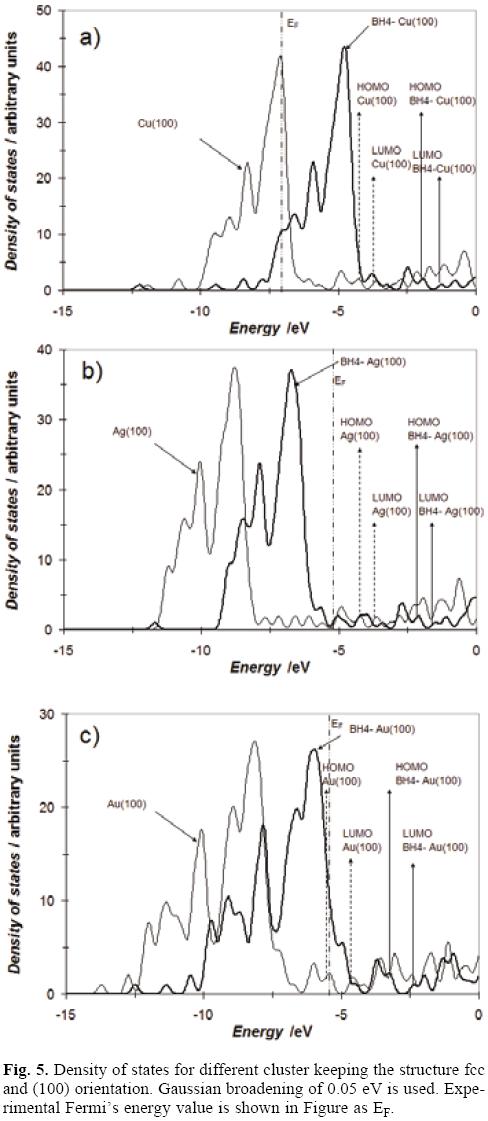
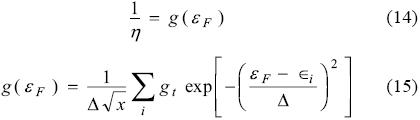
where ∈i are the orbital eigenvalues, gi is the degeneration in the state i, εF is the energy in the Fermi level and Δ is the amplitude of the gaussian function. Here it is important to mention that in systems with few atoms the levels are discrete and the Fermi level corresponds to the HOMO's energy. Under these conditions, from Figure 6, we measured the values of the g(εF) and global hardness associated were 2.3, 6.3 and 11.8 eV for Cu(100), Ag(100) and Au(100) respectively. For BH4--Cu(100), BH4--Ag(100) and BH4--Au(100) the values were 2.5, 2.4 and 2.6 eV respectively. These values compare favorably with those reported in Table 2 and Table 3 suggesting that the models employed in present work are adequate to model the adsorption process of BH4--on Cu(100), Ag(100) and Au(100).
Methodology Models
The monocrystalline surfaces of Cu, Ag an Au with (100) orientation were modeled as finite clusters with face centered cubic (fcc) structure, Figure 6. We used crystallographic parameters for their construction [57]. The orientation (100) was selected because it is the main crystallographic face exhibited by fcc metals [57]. In present work we employed the electronic unit cell (EUC) [15-17]. An EUC is defined as the minimal number of atoms maintaining a structural (crystallographic) pattern that is capable to model a particular electronic property of certain solids or surfaces [15-17]. During the adsorption, we performed restricted optimizations in where the adsorption coordinates of BH4- were optimized while the coordinates of surface atoms were fixed. This procedure was followed because there is not experimental evidence about the existence of a reconstruction process caused by the BH4- ion on the surface where it is adsorbed. The optimal adsorption geometry was obtained employing the hybrid functional B3LYP [58-60] and the relativistic pseudopotentials (LANL2DZ) of Hay and Wadt [61, 62].
Computational resources
We employed a cluster with 32 cores Xeon 3.0 GHZ and 10 GB of memory for all the calculations. These calculations were performed with the package Gaussian 09 [63] and visualized with the GaussView V. 3.05 [64], Gabedit [65] and Gaussum [66] packages.
Conclusions
In present work we performed a theoretical quantum study about the borohydride adsorption onto Cu, Ag and Au at B3LYP/LANL2DZ. The results suggest a more favored adsorption of BH4- on Cu(100) in comparison with Ag(100) or Au(100). Reactivity descriptors such as η y μ and ω involved during the adsorption process of BH4- onto X(100) suggest that gold is a better electrophile. From the condensed Fukui function values it was found that in the electrophilic case the BH4- adsorption increases the reactivity on gold and copper, while deactivates the silver reactivity. For nucleophilic case the copper surface was the most activated compared with silver and gold. For a radical free attack copper and gold showed the higher reactivity.
The evaluations of condensed Fukui function reveled that for the electrophilic case BH4- adsorption activates the silver reactivity, while it deactivate the copper reactivity. For nucleophilic case the gold surface was the most reactive. The spd electronic density of states indicated that the models used reproduce adequately the solid behavior.
Acknowledgments
We acknowledge financial support from the Universidad Autónoma del Estado de Hidalgo through the projects PIFI 2008-13M8U0017T-04-01 y PIFI-2009-13MSU0017T-04-01, and Fondo Mixto CONACyT-Gobierno del Estado de Guanajuato, Grant GTO-2007-C02-69453.
References
1. Dresselhaus, M. S.; Thomas, I. L. Nature 2001, 414, 332-337. [ Links ]
2. Nurul, J. M.; Choudhury, A.; Sahai, Y. Renewable Sustainable Energy Rev. 2010, 14, 183-199. [ Links ]
3. Vielstich, W.; Lamm, A.; Gasteiger, H.A. Handbook of Fuel Cells: Fundamentals, Technology and Applications, Wiley, New York, 2005. [ Links ]
4. Tarasevich, M.R.; Karichev, Z.R.; Bogdanovskaya, V.A.; Lubnin, E.N.; Kapustin, A.V. Electrochem. Commun. 2005, 7, 141-146. [ Links ]
5. de Leon, C.P.; Walsh, F.C.; Pletcher, D.; Browning, D.J.; Lake-man, J.B. J. Power Sources 2006, 155, 172-181. [ Links ]
6. Mirkin, M.V.; Yang, H.; Bard, A.J. J. Electrochem. Soc. 1992, 139, 2212-2217. [ Links ]
7. Chatenet, M.; Micoud, F.; Roche, I.; Chainet, E. Electrochim. Acta 2006, 51, 5459-5467. [ Links ]
8. Rostamikia, G.; Janik, M. J. Electrochim. Acta 2010, 55, 1175-1183. [ Links ]
9. Atwan, M.H.; Macdonald, C.L.B.; Northwood, D.O.; Gyenge, E.L. J. Power Sources 2006, 158, 36-44. [ Links ]
10. Atwan, M.H.; Northwood, D.O.; Gyenge, E.L. Int. J. Hydrogen Energy 2007, 32, 3116-3125. [ Links ]
11. Indig, M.E.; Snyder, R.N. J. Electrochem. Soc. 1962, 109, 11041106. [ Links ]
12. Sanli, E.; Çelikkan, H.; Uysal, B. Z.; Aksu, M. L. Int. J. of Hydrogen Energy 2006, 31, 1920-1924. [ Links ]
13. Demirci, U.B. J. Power Sources 2007, 172, 676-687. [ Links ]
14. Furuya, N.; Koide, S. Surf. Sci. 1989, 220, 18-28. [ Links ]
15. Mendoza-Huizar, L.H.; Palomar-Pardave, M.; Robles, J. J. Mol. Struct. Theochem. 2004, 679, 187-194. [ Links ]
16. Rios-Reyes, C.H.; Camacho Mendoza, R.; Mendoza-Huizar, L.H. J. Mex. Chem. Soc., 2006, 50(1), 19-27. [ Links ]
17. Rios-Reyes, C.H.; Ponce-Rodriguez, A.; Romero-Romo, M.; Mendoza-Huizar, L.H. Rev. Mex. Fis. 2008, 54(2), 104-111. [ Links ]
18. Gázquez, J. L. J. Mex. Chem. Soc. 2008, 52, 3-10. [ Links ]
19. Geerlings, P.; De Proft, F.; Langenaeker, W. Chem. Rev. 2003, 103, 1793-1874. [ Links ]
20. Chermette, H., J. Comput. Chem. 1999, 20, 129-154. [ Links ]
21. Ayers, P.W., Anderson, J.S.M., L.J. Bartolotti, Int. J. Quantum Chem. 2005, 101, 520-534. [ Links ]
22. Chattaraj, P.K., Sarkar, U., Roy, D.R. Chem. Rev. 2006, 106, 2065-2091. [ Links ]
23. Johnson, P.A., Bartolotti, L.J.P., Ayers, W., Fievez, T., Geerlings, P. in Modern Charge Density Analysis, edited by C. Gatti and P. Macchi, Springer, New York, 2012. [ Links ]
24. S.B., Liu, Acta Phys. Chim. Sin. 2009, 25, 590-600. [ Links ]
25. Parr, R.G., Donnelly, R.A., Levy, M., Palke, W.E., J. Chem. Phys. 1978, 68, 3801-3807. [ Links ]
26. Pearson, R.G., Inorg. Chim. Acta 1995, 240, 93-98. [ Links ]
27. Ayers, P.W., Parr, R.G., Pearson, R.G. J. Chem. Phys. 2006, 124, 194107-194121. [ Links ]
28. Ayers, P.W. Faraday Discuss. 2007, 135, 161-190. [ Links ]
29. Parr, R.G.; Szentpaly, L.; Liu, S. J. Am. Chem. Soc. 1999, 121, 1922-1924. [ Links ]
30. Liu, S.B. in Chemical reactivity theory: A density functional view, edited by P.K. Chattaraj, Taylor and Francis, Boca Raton, 2009. [ Links ]
31. Parr, R.G., Yang, W. Density Functional Theory of Atoms and Molecules, Oxford University Press , New York, 1989. [ Links ]
32. Ayers, P. W.; Parr, R.G. J. Am. Chem. Soc. 2000, 122, 2010-2018. [ Links ]
33. Parr, R.G.; Yang, W. J. Am. Chem. Soc. 1984, 106, 4048-4049. [ Links ]
34. Chattaraj, P.K., Lee, H., Parr, R.G. J. Am. Chem. Soc. 1991, 113, 1855-1856. [ Links ]
35. Ayers, P.W. J. Chem. Phys. 2005, 122, 141102-141104. [ Links ]
36. Gazquez, J.L., Mendez, F. J. Phys. Chem. 1994, 98, 4591-4593. [ Links ]
37. Mendez, F., Gazquez, J.L. J. Am. Chem. Soc. 1994, 116, 9298-9301. [ Links ]
38. Parr, R.G., Yang, W.T. J. Am. Chem. Soc. 1984, 106, 4049-4050. [ Links ]
39. Yang, W.T., Parr, R.G., Pucci, R. J. Chem. Phys. 1984, 81, 2862-2863. [ Links ]
40. Ayers, P.W., Levy, M. Theor. Chem. Acc. 2000, 103, 353-360. [ Links ]
41. Ayers, P.W., Yang, W.T., Bartolotti, L.J. in Chemical reactivity theory: A density functional view, edited by P.K. Chattaraj, CRC Press, Boca Raton, 2009. [ Links ]
42. Yang, W.T., Mortier, W.J. J. Am. Chem. Soc. 1986, 108, 5708-5711. [ Links ]
43. Fuentealba, P., Perez, P., Contreras, R. J. Chem. Phys. 2000, 113, 2544-2551. [ Links ]
44. Tiznado, W., Chamorro, E., Contreras, R., Fuentealba, P. J. Phys. Chem. A 2005, 109, 3220-3224. [ Links ]
45. Ayers, P.W., Morrison, R.C., Roy, R.K. J. Chem. Phys. 2002, 116, 8731-8744. [ Links ]
46. Bultinck, P., Fias, S., Alsenoy, C.V., Ayers, P.W., Carbó-Dorca, R. J. Chem. Phys. 2007, 127, 034102-034112. [ Links ]
47. Bultinck, P., Carbo-Dorca, R., Langenaeker, W. J. Chem. Phys. 2003, 118, 4349-4356. [ Links ]
48. Arevalo, R.L. Escaño, M.C.S., Kasa, H. e-J. Surf. Sci. Nanotech. 2011, 9, 257-264 [ Links ]
49. Rostamikia, G., Janik, M.J. Electrochim. Acta 2010, 55, 1175-1183. [ Links ]
50. Rostamikia, G., Janik, M.J. Electrochim. Acta 2010, 55, 1175-1183. [ Links ]
51. G. Rostamikia, A.J. Mendoza, M.A. Hickner, M.J. Janik, J. Power Sources 2011, 196, 9228-923] [ Links ].
52. Parr, R.G.; Pearson, R.G. J. Am. Chem. Soc. 1983, 105, 75127516. [ Links ]
53. Yang, W.T., Parr, R.G. Proc. Natl. Acad. Sci. 1985, 82, 6723-6726. [ Links ]
54. Cohen, M.H., Ganduglia-Pirovano, M.V. J. Chem. Phys. 1994, 101, 8988-8997. [ Links ]
55. Cohen, M.H., Ganduglia-Pirovano, M.V., Kudrnovsky, J. J. Chem. Phys. 1995, 103, 3543-3551. [ Links ]
56. Senet, P. J. Chem. Phys. 1997, 107, 2516-2524. [ Links ]
57. Kittel, C. Introduccion a la Física del Estado Sólido, Reverte, Barcelona, 1976. [ Links ]
58. Becke, A. D. J. Chem. Phys. 1993, 98, 5648-5652. [ Links ]
59. Becke, A. D. Phys. Rev. A 1988, 38, 3098-3100. [ Links ]
60. Lee, C., Yang, W., Parr, R.G. Phys. Rev. B 1988, 37, 785-789. [ Links ]
61. Hay, P.J.; Wadt, W.R. J. Phys. 1985, 82, 270-283. [ Links ]
62. Hay, P.J.; Wadt, W.R. J. Chem. Phys. 1985, 82, 299-310. [ Links ]
63. Gaussian 09, Revision c.01, Frisch, M. J.; Trucks, G. W.; Schlegel, H. B.; Scuseria, G. E.; Robb, M. A.; Cheeseman, J. R.; Montgomery Jr, J. A.; Vreven, T.; Kudin, K. N.; Burant, J. C.; Millam, J. M.; Iyengar, S. S.; Tomasi, J.; Barone, V.; Mennucci, B.; Cossi, M.; Scalmani, G.; Rega, N.; Petersson, G. A.; Nakatsuji, H.; Hada, M.; Ehara, M.; Toyota, K.; Fukuda, R.; Hasegawa, J.; Ishida, M.; Nakajima, T.; Honda, Y.; Kitao, O.; Nakai, H.; Klene, M.; Li, X.; Knox, J. E.; Hratchian, J. P.; Cross, J. B.; Bakken, V.; Adamo, C.; Jaramillo, J.; Gomperts, R.; Stratmann, R. E.; Yazyev, O.; Austin, A. J.; Cammi, R.; Pomelli, C.; Ochterski, J. W.; Ayala, P. Y.; Morokuma, K.; Voth, G. A.; Salvador, P.; Dannenberg, J.J.; Zakrzewski, V.G.; Dapprich, S.; Daniels, A.D.; Strain, M. C.; Farkas, O.; Malick, D. K.; Rabuck, A. D.; Raghavachari, K Foresman, J. B.; Ortiz, J. V.; Cui, Q.; Baboul, A.G.; Clifford, S.; Cioslowski, J.; Stefanov, B. B.; Liu, G.; Liashenko, A.; Piskorz, P.; Komaromi, I.; Martin, R. L.; Fox, D. J.; Keith, T.; Al-Laham, M. A.; Peng, C.Y.; Nanayakkara, A.; Challacombe, M.; Gill, P. M. W.; Johnson, B.; Chen, W.; Wong, M. W.; Gonzalez, C.; Pople, J. A. Gaussian, Inc., Wallingford CT, 2004. [ Links ]
64. Gaussview Rev. 3.09, windows version. Gaussian Inc., Pittsburgh. [ Links ]
65. Allouche, A. R. Gabedit is a free Graphical User Interface for computational chemistry packages. It is available from http://gabedit.sourceforge.net/ [ Links ]
66. Noel M. O'Boyle, Johannes G. Vos, GaussSum 0.9, Dublin City University, 2005. Available at http://gausssum.sourceforge.net [ Links ]
* The asterisk indicates the name of the author to whom inquires about the paper should be addressed.
This paper is dedicated to Professor José Luis Gázquez Mateos for his brilliant contributions to the Density Functional Theory.

