










Servicios Personalizados
Revista
Articulo
 Inglés (pdf)
Inglés (pdf)
 Artículo en XML
Artículo en XML Referencias del artículo
Referencias del artículo
 Enviar artículo por email
Enviar artículo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO -
 Accesos
Accesos
Links relacionados
-
 Similares en
SciELO
Similares en
SciELO
Compartir

 Permalink
PermalinkJournal of the Mexican Chemical Society
versión impresa ISSN 1870-249X
J. Mex. Chem. Soc vol.56 no.3 Ciudad de México jul./sep. 2012
Artículo
The Storage of Hydrogen in Nanoporous Carbons
Julio A. Alonso,1,2* Iván Cabria1 and María J. López1
1 Departamento de Física Teórica, Atómica y Óptica. Facultad de Ciencias, Universidad de Valladolid, E-47011 Valladolid, Spain.
2 Donostia International Physics Center, Paseo M. de Lardizábal, E-20018 San Sebastián, Spain.
Received November 14, 2011.
Accepted March 15, 2012.
Abstract
An efficient storage of hydrogen is a crucial requirement for its use as a fuel in the cars of the future. Experimental and theoretical work has revealed that porous carbons are promising materials for storing molecular hydrogen, adsorbed on the surfaces of the pores. The microstructure of porous carbons is not well known, and we have investigated a class of porous carbons, the carbide-derived carbons, by computer simulation, showing that these materials exhibit a structure of connected pores of nanometric size, with graphitic-like walls. We then apply a thermodynamical model of hydrogen storage in planar and curved pores. The model accounts for the quantum effects of the motion of the molecules in the confining potential of the pores. The optimal pore sizes yielding the highest storage capacities depend mainly on the shape of the pore, and slightly on temperature and pressure. At 300 K and 10 MPa, the optimal widths of the pores lie in the range 6-10 Å. The theoretical predictions are consistent with experiments for activated carbons. The calculated storage capacities of those materials at room temperature fall below the targets. This is a consequence of an insufficiently strong attractive interaction between the hydrogen molecules and the walls of carbon pores. Recent work indicates the beneficial effect of metallic doping of the porous carbons in enhancing the binding energy of H2 to the pore walls, and then the hydrogen storage.
Key words: Carbon; Hydrogen Storage, Porous Materials, Computer Simulation.
Resumen
El posible uso del hidrógeno como combustible en los automóviles del futuro necesita resolver previamente el problema de su almacenamiento. Trabajo experimental y teórico sugiere que los carbones porosos son candidatos prometedores para almacenar hidrógeno, adherido a la superficie interna de los poros. La microestructura de los carbones porosos apenas se conoce. Por ello hemos investigado un tipo de carbones porosos, los carbones derivados de carburos metálicos. Hemos usado técnicas de simulación computacional, mostrando que estos materiales exhiben una estructura formada por poros de tamaño nanométrico interconectados y con paredes grafitizadas. Hemos aplicado un modelo termodinámico para estudiar el almacenamiento de hidrógeno en materiales con poros de paredes planas y poros de paredes curvadas. El modelo tiene en cuenta los efectos cuánticos debido al confinamiento de las moléculas de hidrógeno en los poros. Los tamaños óptimos de poro que permiten las mayores capacidades de almacenamiento de hidrógeno, dependen de la forma de los poros y dependen ligeramente de la temperatura y de la presión a la que se inyecta el hidrógeno. A 300 K y presiones de 10 MPa, las anchuras óptimas de poro están en el intervalo de 6 a 10 Å. Estas predicciones son consistentes con los experimentos en carbones activados. Las capacidades de almacenamiento de estos materiales a temperatura ambiente resultan inferiores a los objetivos actuales. Esto es consecuencia de que la interacción entre las moléculas de hidrógeno y las paredes de los poros no es lo suficientemente atractiva. Trabajos recientes indican que el dopado de los carbones porosos con átomos metálicos y nanopartículas tiene un efecto beneficioso en el proceso de adsorción, aumentando la magnitud de la energía de interacción del H2 con las paredes de los poros y aumentando, en consecuencia, la capacidad de almacenamiento de H2 por estos materiales.
Palabras clave: Carbón, almacenamiento de hidrógeno, materiales porosos, simulación computacional.
Introduction
Hydrogen is expected to play an important role as an alternative fuel to gasoline in the cars of the future [1]. Those cars will use an electric motor based on a hydrogen fuel cell, where the hydrogen gas coming from the storage tank reacts with atmospheric oxygen producing an electric current. The hydrogen cars have advantages over electric battery cars and gasoline cars: hydrogen can be produced from water by electrolysis, and the only emission from the reaction between hydrogen and oxygen is water. In addition, the technology of fuel cells is well developed [2]. Hydrogen has the highest energy content per mass, 120 MJ/kg, compared to other fuels. For instance, the energy content per mass of gasoline is 44 MJ/kg. But hydrogen is a gas, and its energy content per volume is 0.01 MJ/L, much smaller than that of gasoline, 35 MJ/L. Under normal conditions of pressure and temperature, one kg of hydrogen occupies the volume of a football goal. Consequently, the problem is how to store, in a small tank, 5 to 13 kg of hydrogen, depending of the type of vehicle, to cover about 600 km. The short term targets proposed by the US Department of Energy (DOE) in September 2009, which would permit using hydrogen in onboard automotive applications are: a gravimetric storage capacity of 5.5 per cent in weight, and a volumetric capacity of 0.040 kg of hydrogen per litre [3]. The gravimetric capacity of the supporting or adsorbent material, or weight per cent (w %) of stored hydrogen, is defined

where mH is the mass of stored hydrogen, and msupporting is the mass of the supporting material. The volumetric capacity Cv is equal to the mass of stored hydrogen, mH, divided by the volume of the supporting material
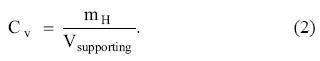
In addition, the storage should be reversible. With these targets fulfilled, hydrogen cars would have the same autonomy range as the present gasoline cars.
In an attempt to reach those targets, different ways to store hydrogen have been investigated. Hydrogen is usually stored as a compressed gas in steel tanks at high pressures of 350-700 bar, and this is the storage technology used in the present hydrogen car prototypes. The main problems are the large volume and weight of the tanks, and the energy spent in compressing hydrogen. Liquid hydrogen occupies much less volume, and is usually stored in cryogenic tanks at temperatures around 20 K. The energy content of liquid hydrogen is high, 8.4 MJ/L. This method provides hydrogen storage capacities of 20 wt % and 0.030 kg/L, but the main disadvantages are the substantial amount of energy spent in the liquefaction of hydrogen, the boil-off losses and the need of superinsulating the tanks. Hydrogen storage in solid materials is a safer method than gas and liquid storage. Two types of promising solid materials are metallic hydrides and porous materials. Metal hydrides store atomic hydrogen dissolved in the crystal lattice of the metal. But the hydrogen atoms are strongly bound to the metal atoms, and a substantial amount of energy has to be spent in releasing the hydrogen. That is, the process is not reversible at room temperature and high temperatures are necessary to release the hydrogen.
There are several types of porous materials that have been studied for storing hydrogen or other gases: porous carbons [4], metal organic frameworks (MOFs) [5, 6], zeolites [7], clathrate hydrates [8], and microporous organic polymers (PIMs) [9]. In all those materials, the relevant mechanism for storing hydrogen is the adsorption of molecular hydrogen on the walls of the pores. The main quantities to determine the amount of hydrogen that can be stored in a material are: the adsorption energy of H2 on the pore walls, the specific surface of the material, that is, the surface area of the pore walls per gram of material, the specific pore volume of the material, that is, the volume contained in the pores per gram of material, and the distribution of pore sizes. Porous materials reach specific surface areas of several thousand m2/g, specific pore volumes in the range 0.11.6 cm3/g, and porosity volumes between 55 and 90%. With respect to the adsorption energy, Eads, of H2 on the surface of the pores, thermodynamic estimations indicate that the values of Eads allowing for an efficient adsorption-desorption cyclic process at room temperature and moderate pressures should be in the range 0.3-0.4 eV/molecule [10]. This is a narrow window in between physisorption and chemisorption. None of the current hydrogen storage methods, and none of the known solid materials meet the DOE targets. Reports of a storage capacity of 5-10 wt % at 273 K and 0.04 MPa in carbon nanotubes [11], have been discarded after more recent experiments [12, 13]. In this article we focus on porous carbons, reviewing our recent work on the storage of hydrogen by this class of materials. The methodology used in the calculations is briefly presented in Section 2. In Section 3 we study the structure of porous carbons by molecular dynamics simulations. Section 4 is dedicated to discuss the interaction of molecular hydrogen with graphitic carbon surfaces. In Section 5 we use a thermodynamic model to present predictions for the hydrogen storage in porous carbons. Section 6 investigates the enhancement of hydrogen storage induced by doping the carbon material, and Section 7 presents the conclusions.
Methodology
The structure of porous carbons is studied in Section 3 by performing molecular dynamics (MD) simulations, with large simulation cells containing more than 60,000 carbon atoms, and long simulation times up to 2500 ps. The interactions between carbon atoms were modeled with the empirical potential developed by Tersoff [14], which represents well the covalent bonding between carbon atoms in diamond and graphite. That potential was combined with a long-range many-body potential due to Nordlund and coworkers [15], representing the weak interaction between layers in graphite. To closely mimic the experimental conditions employed in the production of the class of porous carbons of interest for this work, the simulations have been performed at constant temperature, T, constant number of particles, N, and constant volume V. The interaction between molecular hydrogen and the walls of the pores of clean or doped porous carbons is studied using mainly the density functional formalism [16].
Structure of nanoporous carbons
The term nanoporous carbons is used for a broad class of materials, including activated carbons (ACs) [17], carbide derived carbons (CDCs) [18], schwarzites [19], carbon nanofoams [20], carbon nanotubes [21], carbon nanohorns [22], etc., which exhibit a porous structure with pores of nanometer size. CDCs are produced from inorganic carbides, by the selective extraction of the metal atoms through a chemical chlorination process performed at moderate to high temperatures (873 K to 1473 K). Upon removal of metal atoms from the carbide, the carbon sublattice becomes unstable and the structure collapses internally. It is observed that the material experiences a transformation such that the final pure carbon material has the same macroscopic size and shape as the starting carbide [23]. CDCs have been produced with specific surface areas up to 2300 m2/g, specific pore volumes between 0.2 and 1.0 cm3/g, and porosity volume up to 84 %. The microstructure of the CDCs has been investigated by Raman spectroscopy and high-resolution transmission electron microscopy [24]. At production temperatures near 573 K, the samples look completely amorphous. Near 1073 K, the carbon atoms reorganize to form curved graphene sheets, and at even higher production temperatures, about 1473 K, small pieces of graphite, a few layers thick, develop.
Computational studies of the structure of the ACs and CDCs are recent [25, 26]. To model the porous carbons, Peng and Morris [25] equilibrated liquid carbon at 6000 K, and then quenched the structures to 300 K while keeping the carbon density constant. C-C interactions were modeled with the Tersoff potential [14]. Cells containing 4000 carbon atoms and periodic boundary conditions were utilized in the simulations. We have employed the Tersoff-Nordlund potential [14, 15] to simulate the structure of a nanoporous CDC obtained from a precursor carbide, ZrC [26]. For this purpose, we have performed extensive molecular dynamics (MD) simulations of the collapse of the carbon structure giving rise to the CDC. The macroscopic carbon density, that is, the ratio between the number of atoms and the volume of the simulation cell, remains constant during the simulation, fixed to the experimental value of the carbon density sublattice in zirconium carbide, 0.77 g/cm3. The simulation temperature is the parameter that controls the extent and speed of the structural transformations experienced by the subsystem of carbon atoms once the metallic atoms have been removed from the carbide. Therefore, this temperature is not the same as the chlorination temperature of the experiments, which controls the extraction of the metal atoms from the carbide.
The dynamical simulations begin just after the removal of the metal atoms from the carbide. The initial carbon configuration is taken as a slight distortion of the face-centered-cubic sublattice of the carbon atoms in the carbide. This configuration is unstable and collapses in less than 1 ps. In about 5 ps, small interconnected nanographene ribbons begin to grow, and after 20 ps the main structural characteristics of the CDC are apparent. The material exhibits a structure formed by open interconnected nanopores of different sizes. The pore walls, planar and curved, are formed by one-atom-thick graphitic-like networks of C atoms. However, the pore walls contain many defects. As the simulation proceeds, some defects are annealed out and the nanographene layers forming the pore walls grow larger. Figure 1 shows the results, after 200 ps, of a CDC obtained at a simulation temperature of 3010 K. In this simulation, the cell contains 62,500 atoms, but the figure only shows a depth of 25 Å. The porous structure and the graphitic-like network forming the pore walls are clearly seen. A detail of a typical pore from a simulation at 2100 K is shown in Figure 2, and the large number of defects on the pore walls is apparent.


In a graphene layer, the carbon atoms are arranged in a honeycomb ring pattern, in which the atoms are three-fold coordinated. This structure arises from the sp2 hybridization of the electronic orbitals. Two-fold coordinated atoms are found at the edges of graphene ribbons. Figure 3 shows the percentage of carbon atoms having coordinations one, two, three, and four, as the simulation time progresses. The CDCs contain almost exclusively two-fold and three-fold coordinated atoms. Coordination four is marginal; this indicates the absence of diamond-like carbon. Coordinations two and three develop at the very early stages (in the first 5-10 ps) of the CDC formation. As the time proceeds, the number of carbon atoms with coordination three increases slowly at the expense of those with coordination two. The number of three-fold coordinated atoms increases with the simulation temperature, indicating that the level of graphitization of the pore walls improves. However, even at the highest simulation temperature (T = 3010 K) and for the longest simulation times (2500 ps) there is a 2% fraction of two-fold coordinated atoms. These are the signature of open pore edges.
The ring pattern of the carbon network contains mostly hexagonal rings, but also a sizable fraction of heptagons and octagons (15% of each at T = 2100 K), and a small number (below 2%) of pentagons. The number of hexagons increases with the temperature, confirming the increasing level of graphi-tization of the pore walls. The heptagonal and octagonal rings yield convex surfaces which locally are similar to those found in schwarzites [19]. However, the CDCs lack the long range crystalline order of the schwarzites. The heptagons and octagons are responsible for the open pore structure of the CDCs. Pentagons are required to form closed cage structures, as in the fullerenes, and the small amount of pentagons in the CDCs guarantees that no closed pores are formed. The pore walls exhibit a graphitic-like atomic ordering, but the defects are responsible of the disordered porous structure of the CDCs. In spite of the lack of long range order, in our opinion the CDC should not be considered an amorphous carbon. Amorphous carbons are disordered materials with densities close to that of diamond (3.5 g/cm3). Amorphous carbons contain localized n electrons (in contrast to the aromatic π bonds in graphite) and a varying amount of carbon atoms with sp3 hybridization. In contrast, the CDCs have low densities (about 0.7 g/cm3), and do not contain four-fold coordinated (sp3) atoms.

The quantities used in experiments to characterize nano-porous materials are the specific surface area (SSA) for gas adsorption and the specific pore volume (SPV). The calculated SSA increases with the simulation temperature following the same trend as the experimental SSA with the chlorination temperature. The porosity, i.e., the empty volume of the material, was calculated considering an effective exclusion volume around each atom (volume that cannot be occupied by the adsorbed gas) equal to the volume of a sphere with a radius of 1.7 Å, the van der Waals radius of carbon. The calculated porosity increases from 0.81 cm3/g to 0.87 cm3/g by increasing the simulation temperature between 1000 K and 3000 K. A broader range of variation of the specific pore volume (0.55-0.90 cm3/g) is obtained in the experiments for chlorination temperatures ranging between 1000 K and 1500 K. However, very similar largest specific pore volumes are obtained in the simulations and in the experiments.
Nanoporous materials are not fully characterized by their SSA and SPV [17]. The hydrogen adsorption capacity depends also on the size of the pores [27]. Optimal pore sizes of 7-11 Å have been predicted for adsorption at room temperature [28, 29]. A detailed characterization of the porous structure of a material is given through the pore size distribution function (PSDF). To obtain meaningful distribution functions, one has to count a sufficiently large number of pores within the sample, which requires very large simulation cells. For this purpose we have used simulation cells containing 62500 carbon atoms. We found that the average size of the pores increases from 0.94 nm to 1.43 nm when the simulation temperature increases between 350 K and 3010 K. At the same time, the pore size distribution becomes broader. The experimental PSDFs of CDCs from ZrC precursor exhibit an enhancement of the average pore size, from 0.80 to 1.41 nm, for chlorination temperatures ranging from 900 K to 1500 K, and a broadening of the pore size distribution [27]; a good correlation exists [26] between simulation and experiment.
In summary, simulations of the CDCs reveal the formation of interconnected open pore structures with graphene-like walls. The pore walls exhibit a substantial degree of graphitization which increases with the annealing temperature. However, a number of defects are present in the carbon network.
Interaction of molecular hydrogen with carbon surfaces
The mechanism underlying the potential of porous carbons to store hydrogen is the physisorption of molecular hydrogen on the graphitic pore walls. Li et al. [10] have estimated that the adsorption enthalpy for the occurrence of reversible adsorption/desorption under the conditions required for automotive applications, which are room temperature and moderate pressures, should be near 0.3-0.4 eV/molecule. Three calculated interaction potential curves of a hydrogen molecule with a graphene layer are shown in Figure 4. The density functional (DFT-LDA) potential [28] captures the weak interaction between the molecule and the graphene layer, although overestimating its strength. However, the LDA fails in representing the long range behavior of the potential, because it does not include dispersion forces. The Lennard-Jones (LJ) potential of Bhatia and Myers [30] gives, by construction, the correct behavior at long distances, but it yields a too weak interaction at equilibrium compared to the other two potentials. The strength and equilibrium distance of the Möller-Plesset (MP2) interaction potential are intermediate between those of the LJ and the DFT-LDA potentials. In those MP2 calculations [31], the graphene layer was simulated by a planar coronene, C24H12, with the dangling bonds saturated by hydrogen atoms. The MP2 method provides a reliable account of dispersion forces, and the interaction potential is more accurate than the other two plotted in Fig. 4. The adsorption energy, Ead, amounts to 0.064 eV/molecule in the MP2 calculation. Ead is defined

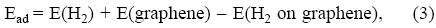
where E(H2) is the energy of the free hydrogen molecule, E(graphene) is the energy of the clean graphene layer, and E(H2 on graphene) is the energy of the combined system formed by the graphene layer with the hydrogen molecule adsorbed in its equilibrium configuration. In a general framework, the repulsive wall of the potential is due to the short-range repulsion that develops when the closed electronic shell of the hydrogen molecule overlaps substantially with the electron density of the substrate. The attractive contribution is due, in part, to the non-linearity of the exchange-correlation effects, and to the small relaxation of the electron density for H2-graphene distances near the minimum of the potential [32]. The van der Waals forces, which can be viewed as arising from nonlocal electronic correlations [33], contribute the rest of the attractive part near the minimum, and the whole of the long range behavior.
It is clear from Fig. 4 that the energy of adsorption of molecular hydrogen on the surface of a graphitic nanostructure is lower than 0.10 eV/molecule. LDA calculations for H2 adsorbed on the outer wall of carbon nanotubes led to the same conclusion [34, 35]. These binding energies fall short of the value required for reversible adsorption/desorption at room temperature and normal pressures. The small adsorption energies explain why the measured hydrogen storage capacities of carbon nanotubes at room temperature are less than 1 wt % [12] (in the experiments, hydrogen is mostly adsorbed on the outer surface of the nanotubes).
The adsorption energy is enhanced when the hydrogen molecules are confined in between two closely spaced parallel surfaces, or near a concave surface. This is achieved inside nanoporous materials [24, 27, 36, 37]. A slitpore, consisting of two parallel graphene layers separated by a certain distance d, gives a simple model for the pores existing in nanoporous carbons. The measured gas adsorption properties of nanoporous carbons are usually analyzed by assuming the pores to have the form of slitpores. The minimum of the interaction potential shown in Fig. 4 occurs for a distance close to 3 Å, so in a slit-pore having a width d of about 6 Å the hydrogen molecule interacts with the two graphene walls, increasing the binding energy by factor of two. The adsorption energy also increases inside a cylindrical nanopore: adsorption energies inside (5, 5) and (6, 6) nanotubes obtained in DFT-LDA calculations are 0.12 eV/molecule and 0.17 eV/molecule, respectively, compared to a value of 0.07 eV/molecule for adsorption on the outer surface [34]. The adsorption energy inside a pore with the form of a half-fullerene reaches the value Ead = 0.2 eV/molecule [37].
Hydrogen storage in porous carbons
The predictions of enhanced adsorption energies in pores of different forms are encouraging and have motivated calculations of the gravimetric and volumetric hydrogen storage capacities for model porous materials. A thermodynamic method has been applied to predict the extent of hydrogen storage in materials formed by graphene slitpores [28, 29, 38]. That method takes into account the quantum behavior of the molecules confined in the pores. The amount of hydrogen adsorbed on the pore walls at a given temperature T is determined by the thermodynamic equilibrium between the adsorbed hydrogen phase and a non-adsorbed compressed gas phase.
As a first step, the quantum states with energies εi for the motion of the hydrogen molecule in the potential v(r) of the pore are calculated by solving the time-independent Schroding-er equation of quantum mechanics. Figure 5 shows the confining potential for slitpores of width d = 6.6 Å and d = 10 Å. This potential is v(z) = v1(z) + v2(d-z), where v1(z) and v2(d-z) represent the interaction with the left-side and right-side walls, respectively, as given by the MP2 curve of Figure 4. This is the origin of the two minima for d = 10 Å. In the thinner pore, only one deep minimum appears. The horizontal lines represent the energies εi of the quantum states of the molecule, obtained by solving the Schrodinger equation for the motion of a particle of mass m = m(H2) in the pore potential v(z). With the energy eigenvalues ei one evaluates the partition function of the adsorbed hydrogen phase,  , where T is the temperature and k is Boltzmann's constant. In practice, only the two or three deepest states contribute to Zad. The corresponding partition function of non-adsorbed (free) molecules confined in the volume of the pore is
, where T is the temperature and k is Boltzmann's constant. In practice, only the two or three deepest states contribute to Zad. The corresponding partition function of non-adsorbed (free) molecules confined in the volume of the pore is  , where h is Planck's constant. The quantity dexcl is an exclusion distance accounting for the strong repulsion of the interaction potential very close to the pore wall. This steep repulsion is clearly appreciated in Figs. 4 and 5. The ratio Keq = Zad/Zfree gives the equilibrium constant between the adsorbed and the compressed phases. Then Keq allows relating the external applied pressure Pext and the pressure of the adsorbed phase, also called internal pressure, Pint, through the equation:
, where h is Planck's constant. The quantity dexcl is an exclusion distance accounting for the strong repulsion of the interaction potential very close to the pore wall. This steep repulsion is clearly appreciated in Figs. 4 and 5. The ratio Keq = Zad/Zfree gives the equilibrium constant between the adsorbed and the compressed phases. Then Keq allows relating the external applied pressure Pext and the pressure of the adsorbed phase, also called internal pressure, Pint, through the equation:


where R is the gas constant. A key ingredient in this equation is the molar volume Vmolar(P,T), which is given by the equation of state, EOS, of hydrogen. Starting from the experimental information available [39, 40], we have built an accurate parameterization of the EOS of hydrogen [28, 29]. From Eq. (4) one obtains Pint for given values of Pext and T, and finally the molar volume of the adsorbed hydrogen phase, Vmol (Pint, T), is obtained from the EOS. In the experiments, the stored hydrogen is composed of two phases, adsorbed hydrogen and compressed gas. The excess storage capacity, due to the adsorbed phase only, is the relevant one.
Figure 6 shows the predictions for the gravimetric and volumetric storage capacities of a carbon material formed by slitpores, at T = 300 K and external pressure of 10 MPa. Results are presented for the three potentials, LJ, MP2 and DFT. The qualitative behaviour of the storage capacities as a function of pore width is the same. However, there are quantitative differences. The largest differences occur in the region around the maxima of the storage capacities, for pore widths in the interval 5-10 Å. The DFT storage capacities are larger than the MP2 capacities, and these are larger than the LJ capacities. This trend correlates with the depth of the DFT, MP2 and LJ potentials in Figure 4. It is interesting to notice the agreement in predicting maximum storage for pores with sizes between 6 and 7 Å. The optimal pore sizes are in good agreement with experiments and previous theoretical results. Rzepka et al. [41] made grand canonical Monte Carlo (GCMC) simulations for the storage of molecular hydrogen in slitpores, obtaining doptimum ≈ 7 A at 300 K and 10 MPa. Wang and Johnson performed GCMC path integral simulations, obtaining optimal pore widths of 6 and 9 Å at 288 and 77K, respectively [42]. De la Casa-Lillo et al. [43] reported an experimental pore width, doptimum = 6.6 Å at 293 K and 70 MPa. This pore size was obtained from the measured curves of the volumetric capacity in activated carbons and activated carbon fibers. The storage capacities read from Figure 6 are below the DOE target. For instance, at the optimized pore size, the gravimetric capacity w(MP2) is equal to 0.8 wt %, and the volumetric capacity Cv is 0.018 kg/L. The predicted storage capacities increase substantially at low temperature [28, 29]. At 77 K, the optimum pore width increases to 9-10 Å, and w reaches values of more than 3 wt %. Cv becomes equal to 0.06 kg/L, which fulfills the DOE objective. However, those low temperatures have no practical interest, except in outer space vehicles. In our model, each graphene wall is part of a single pore. In a previous publication [28] we assumed each wall to be part of two adjacent pores. This increases w by a factor of 2. The situation in real materials is in between. A factor 1.5 is probably realistic.

Pores in carbon materials can have different forms, resembling not only slitpores, but also open cylindrical or near spherical pores. Calculations of the hydrogen storage of those pores have also been performed [29, 37]. Figure 7 compares the calculated storage capacities of planar, cylindrical and spherical pores at 300 K and 10 MPa. The storage capacities of the slit-pores are the highest, although the differences are not large.

Enhancement of hydrogen storage by metal doping
Doping of carbon materials with metal impurities or small metal clusters represents a promising strategy to enhance the binding of molecular hydrogen to the walls of the pores. An increase in the storage capacities of Li-doped and K-doped carbon nanotubes, as compared to clean nanotubes, has been observed [44, 45]. An atypically large hydrogen uptake on chemically-activated porous carbon has been proposed to be due to traces of alkali metals residual from chemical activation [46]. Doping with transition metal impurities also looks promising [47].
The most stable position of a Li atom adsorbed on a graphene layer is above the center of a carbon hexagon. The adsorption energy of the Li atom in DFT calculations is close to 1 eV, and the equilibrium distance between the Li atom and the carbon layer is 1.7-1.8 Å [48, 49]. The first panel of Figure 8 shows the electron density redistribution


The yellow and brown color regions above the Li atom represent a loss of electron density (negative Δρ), and the transferred charge concentrates in the region (green and blue colors) between Li and the carbon layer. The net electron transfer is 0.5-0.6 e [48, 50, 51]. The Li-graphene interaction is partly ionic and partly covalent.
Cabria et al. [48] and Cho and Park [52] have performed LDA and GGA (generalized gradient approximation) calculations, respectively, comparing the adsorption of H2 on pure and Li-doped graphene and nanotubes. Figure 9 shows a comparison of the interaction potential of H2 with clean and Li-doped graphene in LDA calculations. The interaction potential with the Li-doped graphene is more attractive. The adsorption energy on the Li-doped graphene is given by

Ead-doped = E(H2) + E(Li-doped graphene) -
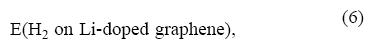
where E(H2 on Li-doped graphene) is the energy of the system at the minimum of the interaction potential of Figure 9. Thus, the DFT H2 adsorption energies are enhanced substantially by the presence of lithium, reaching values of 0.17-0.20 eV per molecule, which represent an increase by a factor of two with respect to the adsorption energies on clean graphene. A similar enhancement is found in calculations for adsorption of H2 on nanotubes doped with lithium [48, 52] and potassium [53]. It is reasonable to expect that the presence of Li will induce a similar increase, by a factor of two, in the MP2 H2-graphene interaction potential, whose depth could then be estimated as 0.12-0.13 eV. The origin of the enhancement of the adsorption energy is the polarization of the charge of the H2 molecule by the charged alkali atoms. The second panel of Figure 8 shows the polarization of the charge of the hydrogen molecule due to the presence of Li. A rough estimate can be made of the effect of Li doping on the hydrogen storage capacity. It has been mentioned in Section 5 that the calculated maximum storage capacities correlate with the depth of the interaction potentials. This correlation is nearly linear. Using that correlation, we find that the MP2 H2-graphene interaction potential, corrected by the presence of lithium, leads to storage capacities near 1.5 wt % in slitpores. This value may increase to 2 wt % if we allow for the fact that many carbon walls form part of two adjacent pores.
The combined effect of doping and confinement in a narrow curved pore has been studied [37]. LDA calculations for H2 adsorption on the inner surface of a pore with bowl-like shape doped with Li yield adsorption energies in the range 0.28-0.33 eV/molecule. Although a bit overestimated due to the use of the LDA, these are very encouraging values which support the promising effect of metal doping. The calculations of Sigal and coworkers [54] have shown that oxygen interferes with hydrogen adsorption in the doped carbons, by either blocking the adsorption sites or by the oxidation of the metal decoration. Consequently, it is necessary to reduce the oxygen partial pressure sufficiently to allow for hydrogen storage.
Conclusions
An efficient storage of hydrogen is a crucial requirement for its use as a fuel in the cars of the future. Those cars will use hydrogen fuel cells instead of electric batteries or gasoline engines, releasing only water. Nanoporous carbons are promising materials for storing hydrogen, adsorbed in molecular form on the inner surfaces of the pores. Because of their porous structure, these materials have an enormous specific surface area. The microstructure of porous carbons at an atomic scale is not well known and we have investigated a class of porous carbons, the carbide-derived carbons, or CDCs. The simulations have shown that the CDCs exhibit a structure of interconnected nanopores with graphitic-like walls.
Using a quantum-thermodynamic model and H2-carbon wall interaction potentials derived from different theoretical and empirical approaches we have studied the storage of hydrogen in carbon materials with pores of different sizes and shapes. Although the calculated storage capacity of those materials at cryogenic temperatures is sizable, at room temperature the storage capacities fall below the DOE targets, indicating that just an optimization of the pore size and shape is not enough to reach those targets. This is a consequence of an insufficiently strong attractive interaction between the hydrogen molecule and the walls of carbon pores. Recent work indicates the beneficial effect of some metallic impurities in enhancing the binding energy of H2 to the pore walls. Consequently, nanoporous carbons doped with light metallic impurities like lithium appear as promising candidates for this task. The effect of heavier dopants, like palladium and other transition metals, is also a topic of current interest.
Acknowledgments
We dedicate this paper to Professor José Luis Gázquez. Work supported by MEC of Spain and the European Regional Development Fund (Grants MAT2008-06843-C03-01 and MAT2011-22781), and Junta de Castilla y León (Grant VA158A11-2). JAA acknowledges the hospitality and support of the Donostia International Physics Center.
References
1. Ogden, J. M. Phys. Today, 2002, 55(4), 69. [ Links ]
2. EG&G Technical Services, Inc., Fuel Cell Handbook (Seventh Edition), US Department of Energy, Morgantown 2004. [ Links ]
3. Office of Energy Efficiency and Renewable Energy and The FreedomCAR and Fuel Partnership, Targets for onboard hydrogen storage systems for light-duty vehicles. US Department of Energy, September 2009. http://www.eere.energy.gov/hydrogenandfuelcells/storage/pdfs/targets_onboard_hydro_storage_explanation.pdf. [ Links ]
4. Cabria, I.; López, M. J.; Alonso, J. A. Handbook of Nanophysics, Vol. 5: Functional Materials. Editor K. Sattler, CRC Press, Boca Raton 2010, p. 41.1 [ Links ]
5. Wong-Foy, A. G.; Matzger, A. J.; Yaghi, O. M. J. Am. Chem. Soc. 2006, 128, 3494. [ Links ]
6. Cabria, I.; López, M. J.; Alonso, J. A. Phys. Rev. B 2008, 78, 295324. [ Links ]
7. Langmi, H. W.; Walton, A.; Al-Mamouri, M. M.; Johnson, S. R.; Book, D.; Speight, J. D.; Edwards, P. P.; Gameson, I.; Anderson, P. A.; Harris, I. R. J. Alloys Comp. 2003, 356, 710. [ Links ]
8. Mao, W. L.; Mao, H. K.; Goncharov, A. F.; Struzhkin, V. V.; Guo, Q.; Hu, Shu, J.; Hemley, R. J.; Somayazulu, M.; Zhao, Y. Science 2002, 297, 2247. [ Links ]
9. McKeown, N. B.; Budd, P. M.; Book, D. Macromol. Rapid Commun. 2007, 28, 995. [ Links ]
10. Li, J.; Furuta, T.; Goto, H.; Ohashi, T.; Fujiwara, Y.; Yip, S. J. Chem. Phys. 2003, 119, 2376. [ Links ]
11. Dillon, A. C.; Jones, K. M.; Bekkedahl, T. A.; Kiang, C. H.; Bethune, D. S.; Heben, M. J. Nature 1997, 386, 377. [ Links ]
12. Züttel, A. Mater. Today 2003, 6, 24. [ Links ]
13. Ansón, A.; Callejas, M. A.; Benito, A. M.; Maser, W. K.; Izquierdo, M. T.; Rubio, B.; Jagiello, J.; Thommes, M.; Parra, J. B.; Martinez, M. T. Carbon 2004, 42, 1243. [ Links ]
14. Tersoff, J. Phys. Rev. Lett. 1988, 61, 2879. [ Links ]
15. Nordlund, K.; Keinonen, J.; Mattila, T. Phys. Rev. Lett. 1996, 77, 699. [ Links ]
16. Dreizler, R. M.; Gross, E. K. U. Density Functional Theory. Springer, Berlin 1990. [ Links ]
17. Linares-Solano, A.; Jordá-Beneyto, M.; Kunowsky, M.; Suárez-García, F.; Cazorla-Amorós, D. Carbon Materials: Theory and Practice. Editors P. Terzyk, P. A. Gauden and P. Kowalczyk, Research Signpost, Trivandrum 2008, p. 245. [ Links ]
18. Dash, R. K.; Yushin, G.; Gogotsi, Y. Microporous Mesoporous Mater. 2005, 86, 50. [ Links ]
19. Lenosky, T.; Gonze, X.; Teter, M.; Elser, V. Nature 1992, 355, 333. [ Links ]
20. Barborini, E.; Piseri, P.; Milani, P.; Benedek, G.; Ducati, C.; Robertson, J. Appl. Phys. Lett. 2002, 81, 3359. [ Links ]
21. Carbon Nanotubes. Synthesis, Structure, Properties, and Applications, Editors M. S. Dresselhaus, G. Dresselhaus and Ph. Avouris. Springer-Verlag, Berlin 2001. [ Links ]
22. Iijima, S.; Yudasaka, M.; Yamada, R.; Bandow, S.; Suenaga, K.; Kokai, F.; Takahashi, K. Chem. Phys. Lett. 1999, 309, 165. [ Links ]
23. Gogotsi, Y.; Nikitin, A.; Ye, H.; Zhou, W.; Fischer, J. E.; Yi, B.; Foley, H. C.; Barsoum, M. W. Nature Mater. 2003, 2, 591. [ Links ]
24. Yushin, G.; Dash, R.; Jagiello, J.; Gogotsi, Y. Adv. Funct. Mater. 2006, 16, 2288. [ Links ]
25. Peng, L.; Morris, J. R. J. Phys. Chem. C 2010, 114,15522. [ Links ]
26. López, M. J.; Cabria, I.; Alonso, J. A. J. Chem. Phys. 2011, 135, 104706. [ Links ]
27. Gogotsi, Y.; Dash, R. K.; Yushin, G.; Yildirim, T.; Laudisio, G.; Fischer, J. E. J. Am. Chem. Soc. 2005, 127, 16006. [ Links ]
28. Cabria, I.; López, M. J.; Alonso, J. A.; Carbon 2007, 45, 2649. [ Links ]
29. Cabria, I.; López, M. J.; Alonso, J. A. Int. J. Hydrogen Energy 2011, 36, 10748. [ Links ]
30. Bhatia, S. K.; Myers, A. L. Langmuir 2006, 22, 1688. [ Links ]
31. Ferre-Vilaplana, A. J. Chem Phys. 2005, 122, 104709. [ Links ]
32. Arellano, J. S.; Molina, L. M.; Rubio, A.; Alonso, J. A. J. Chem. Phys. 2000, 112, 8114. [ Links ]
33. Alonso, J. A.; Mañanes, A. Theor. Chem. Accounts, 2007, 117, 467. [ Links ]
34. Arellano, J. S.; Molina, L. M.; Rubio, A.; López, M. J.; Alonso, J. A. J Chem. Phys. 2002, 117, 2281. [ Links ]
35. Cabria, I.; López, M. J.; Alonso, J. A. Comput. Mater. Sci. 2006, 35, 238. [ Links ]
36. Jorda-Beneyto, M.; Suárez-García, F.; Lozano-Castelló, D.; Cazorla-Amorós, D.; Linares-Solano, A. Carbon 2007, 45, 293. [ Links ]
37. Cabria, I.; López, M. J.; Alonso, J. A. J. Chem. Phys. 2008, 128, 144704. [ Links ]
38. Patchkovskii, S.; Tse, J. S.; Yurchenko, S. N.; Zhechkov, L.; Hrine, T.; Seifert, G. Proc. Natl. Acad. Sci. USA 2005, 102,10439. [ Links ]
39. Mills, R. L.; Liebenberg, D. H.; Bronson, J. C.; Schmidt, L. C. J. Chem. Phys. 1977, 66, 3076. [ Links ]
40. Younglove, B. A. J. Phys. Chem. Ref. Data 1982, 11, 1. [ Links ]
41. Rzepka, M.; Lamp, P.; de la Casa-Lillo, M. A. J. Phys. Chem. B 1998, 102, 10894. [ Links ]
42. Wang, Q.; Johnson, J. K. J. Chem. Phys. 1999, 110, 577. [ Links ]
43. de la Casa-Lillo, M. A.; Lamari-Darkrim, F.; Cazorla-Amorós, D.; Linares-Solano, A. J. Phys. Chem. B 2002, 106, 10930. [ Links ]
44. Chen, P.; Wu, X.; Lin, J.; Tan, K. L. Science 1999, 285, 91. [ Links ]
45. Yang, R. T. Carbon 2000, 38, 623. [ Links ]
46. Bath, V. V.; Contescu, C. I.; Gallego, N. C.; Baker, F. S. Carbon 2010, 48, 1331. [ Links ]
47. Contescu, C. I.; van Benthem, K.; Li, S.; Bonifacio, C. S.; Pennycook, S. J.; Jena, P.; Gallego, N. C. Carbon 2011, 49, 4050. [ Links ]
48. Cabria, I.; López, M. J.; Alonso, J. A. J. Chem. Phys. 2005, 123, 204721. [ Links ]
49. Chan, K. T.; Neaton, J. B.; Cohen, M. L. Phys. Rev. B 2008, 77, 235430. [ Links ]
50. Khantha, M.; Cordero, N. A.; Molina, L. M.; Alonso, J. A.; Girifalco, L. A. Phys. Rev. B 2004, 70, 125422. [ Links ]
51. Zhu, Z. H.; Lu, G. Q. Langmuir 2004, 20, 10751. [ Links ]
52. Cho, J. H.; Park, C. R. Catal. Today 2007, 120, 407. [ Links ]
53. Froudakis, G. E. Nano Lett. 2001, 1, 531. [ Links ]
54. Sigal, A.; Rojas, M. I.; Leiva, E. P. M. Phys. Rev. Lett. 2011, 107, 158701. [ Links ]
* The asterisk indicates the name of the author to whom inquires about the paper should be addressed.

