










Servicios Personalizados
Revista
Articulo
 Inglés (pdf)
Inglés (pdf)
 Artículo en XML
Artículo en XML Referencias del artículo
Referencias del artículo
 Enviar artículo por email
Enviar artículo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO -
 Accesos
Accesos
Links relacionados
-
 Similares en
SciELO
Similares en
SciELO
Compartir

 Permalink
PermalinkJournal of the Mexican Chemical Society
versión impresa ISSN 1870-249X
J. Mex. Chem. Soc vol.56 no.3 Ciudad de México jul./sep. 2012
Artículo
Do Casiopeinas® Prevent Cancer Disease by Acting as Antiradicals? A Chemical Reactivity Study Applying Density Functional Theory
Mayra Avelar and Ana Martínez*
Instituto de Investigaciones en Materiales, Universidad Nacional Autónoma de México, Circuito Exterior, Ciudad Universitaria, México 04510, D.F. martina@iim.unam.rnx
Received November 29, 2011.
Accepted March 06, 2012.
Abstract
The main goal of this investigation is to study the possible mechanisms of Casiopeinas® as anticancer agents. Electrodonating (χ-) and electroaccepting (χ+) electronegativity were calculated applying Density Functional Theory. Two different anticancer mechanisms of Casiopeínas® are proposed. There might be antiradical molecules preventing the formation of cancer cells or these molecules could reduce the amount of GSH and as a result over-produce free radicals, increasing the oxidative stress which in turn kills the cancer cells.
Key words: Copper Compounds, Casiopeinas®, Electrodonating, Electroaccepting, Electronegativity, Anticancer, Free Radicals.
Resumen
El principal objetivo de esta investigación es analizar los posibles mecanismos de reacción de las Casiopeinas® como agentes anticancerígenos. Para esto se calculó la electronegatividad como electrodonador (χ-) y como electroaceptor (χ+) utilizando la Teoría de Funcionales de la Densidad. Se proponen dos mecanismos: las Casiopeinas® como antirradicales previenen la formación de células cancerosas, o pueden reducir la cantidad de GSH y con eso provocar una sobreproducción de radicales libres, incrementando el estrés oxidativo que acaba con las células cancerosas.
Palabras clave: Compuestos de cobre, Casiopeinas®, electrodonador, electroaceptor, electronegatividad, anticáncer, radicales libres.
Introduction
Density Functional Theory (DFT) [1-5] of chemical reactivity was born three decades ago and has provided a solid framework for the study of the reaction mechanism operating in a number of processes. The starting point of chemical reactivity theory can be identified with the definitions of fundamental concepts such as electronegativity, hardness and the hard and soft acid base theory reported by Parr, Pearson, Donelly, Levy and Palke [6, 7]. These concepts were previously established intuitively but were not useful for determining absolute values or for understanding associated principles. Taking the solid definitions derived from DFT, it is possible to quantify these parameters and to make a quantitative prediction of the chemical reactivity inherent in many molecules. In the light of ideas put forward by Parr et al., other new concepts contributing very important information about the response of a chemical species when it reacts with other chemical reagents were defined by several authors, including Gazquez [8-17]. There are global and local parameters which permit us to identify the most relevant aspects of the interaction between two chemical species. These parameters consist of response functions that describe how the electronic structure of an isolated species is affected by the presence of another molecule. Within this theory, electron transfer can be considered as an initial stage in the interaction between two molecules when they are separate from each other. The response functions are defined in terms of the derivatives of the total energy with respect to the number of electrons. The chemical potential (μ) and the global hardness (η) are global parameters which are equal to the first and second derivative (respectively) of the total energy, with respect to the number of electrons, with constant external potential. Chemical potential refers to the measurement of charge flow directions and chemical hardness evaluates resistance to the flow of electrons. A simple charge-transfer model of a molecule immersed in an idealized environment that may either withdraw or donate charge was analysed as the global response to the electron transfer process. The evaluation of the capacity of a molecule to either accept or donate charge is achieved by a quadratic interpolation for the energy as a function of the number of electrons. Following on from this definition, the response of a molecule to charge acceptance or withdrawal is calculated in terms of vertical electron affinity (A) and vertical ionization potential (I). Within this approximation, Gazquez and co-workers [15-17] detailed two different chemical potentials, making it possible to distinguish the response to charge donation (μ-) from the response to charge acceptance (μ+). Since the additive inverse of chemical potential is electronegativity (χ), from these parameters it is possible to define two different electronegativities for the charge transfer process: one indicating donation (χ-) and the other indicating electron acceptance (χ+)
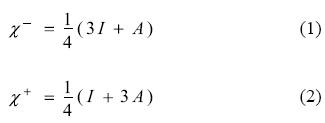
Lower values for χ- imply a more effective electron donor and higher values for χ+ indicate greater capacity for accepting electrons. These and other parameters were used in the past to evaluate the electron transfer process, as the first step in the reaction of several molecules, and also to evaluate the capacity of many substances to prevent the oxidative stress [18-27].
One mechanism for preventing oxidative stress discussed in the literature is the electron transfer reaction between free radicals and free radical scavengers. It was previously reported that in order to scavenge free radicals, molecules can either donate or accept unpaired electrons [18, 28-44]. The antiradical reaction inhibits the damage of the free radical species, either by causing its oxidation or by causing its reduction. An antioxidant is oxidized by transferring electrons to the free radical and preventing other molecules from undergoing oxidation with the free radical. This reaction halts oxidation and for this reason these molecules can be clearly identified as antioxidants. On the other hand, an antireductant is reduced by accepting electrons from the free radical. This process prevents the reduction of other molecules. Since the relevant event is the trapping of free radicals, it would be more precise to refer to these substances as "antiradicals". Antioxidation and antireduction represent two sides of the same coin: antiradical activity through electron transfer.
Antiradicals are important, not only because they prevent oxidative stress, but also because they may help to prevent cancer disease. It was previously reported that free radicals are able to develop cancer damaging DNA by stealing electrons. Following this idea, many investigations were undertaken in order to find evidence concerning the apparent reduction of cancer risk derived by consuming antioxidants [45-48]. Although evidence exists which supports this idea, the protective effect provided by antioxidants is still unproven. It is not possible to conclude with certainty that antioxidants effectively prevent cancer, because apparently both the nature of the molecule acting as free radical scavenger is important, as well as the concentration of these substances. There is also evidence that some antioxidant effects may be related to the induction of oxidative stress. Apparently, pro-oxidative effects may be related to the induction of apoptosis in tumour cells. If this is the case, both the electron donor and the electron acceptor capacity of free radical scavengers turn out to be important.
For over two decades [50-58], new antineoplastic and genotoxic compounds were synthesized and reported. These substances are metal-based drugs named Casiopeinas® and they are a series of mixed chelate copper complexes which are being evaluated as possible anticancer agents. In particular, the activity of casiopeina II [Cu(1,4-dimethyl-1,10-phenanthroline) (glycine)NO3] was tested in cells where apparently this copper complex kills cells by apoptosis and necrosis. The effects of these compounds on the cell include oxidative damage and mi-tochondrial dysfunction. However, the molecular mechanism leading to such effects remains unclear. It was suggested that Casiopeinas® might alter levels of cellular 7-glutamyl-cyste-inyl-glicine (GSH, also known as tripeptide glutathione) by redox reactions which both generate and cause the over-production of free radical species, leading to mitochondrial dysfunction and cell death. Although some experimental reports exist which describe these reactions, neither the mechanism nor the role of Casiopeinas® as electron donors or acceptors is clear. For this reason, the aim of this investigation is to characterize mixed chelate copper complexes through the DFT of chemical reactivity parameters. Thus we will demonstrate that Casiopeinas® are able to accept electrons from the superoxide anion and also from the GSH molecule in its reduced form. The hypothesis of this work is that Casiopeinas® are able to act as antiradicals and thus negate the effect of free radicals on the development of cancer by preventing damage to DNA caused by redox reactions, likewise accepting electrons from GSH in its reduced form and thus affecting the free radical scavenging capacity of this molecule and increasing the concentration of free radicals. To decide which of these two mechanisms prevails over the other is an area requiring investigation.
Results and Discussion
In order to study the parameters of reactivity which might aid in understanding the characteristics of Casiopeinas®, nine different chelate copper complexes were investigated using Density Functional Theory calculations. These molecules have the following condensed formulae: [Cu(N-N)(O-N)]+1 and [Cu(N-N)(O-O)]+1. For the (N-N) ligands we used four different molecules: 1,10-phenathroline (phen), 4,7-dimethyl, 1,10-phenan-throline (dmphen), 2,2'-bipyridine (bipy), and 4,4'-dimethyl, 2,2'-bipyridine (dmbipy). The (O-N) molecules are two different aminoacidates: glycinato (gly) and alaninato (ala). The (O-O) molecule that was used is acetylacetonato (acac). The schematic representation of the studied compounds is shown in Figure 1. Optimized geometries, selected bond distances and atomic charges (APT) [68] are presented in Table 1. The theoretical bond lengths concur well with experimental values. The Cu-N bond distances are greater than the Cu-O bond lengths. The longest distance is that between the Cu atom and the oxygen atom of the water molecule at the apical position. The Cu atom is at the center of a square pyramid and the base of the pyramid is almost planar. In all the compounds, the Cu atom is positive and the most negative atom is the oxygen of the (O-N) or the (O-O) molecules.
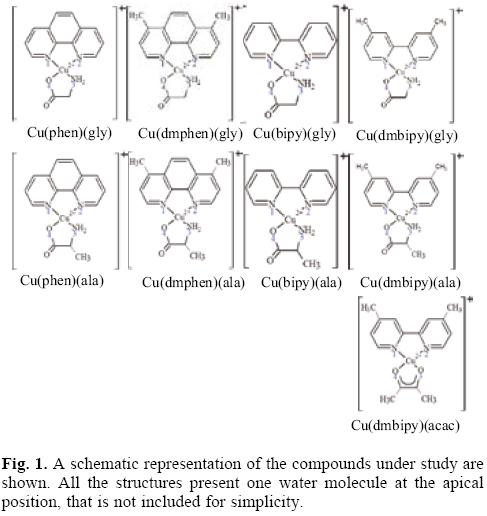
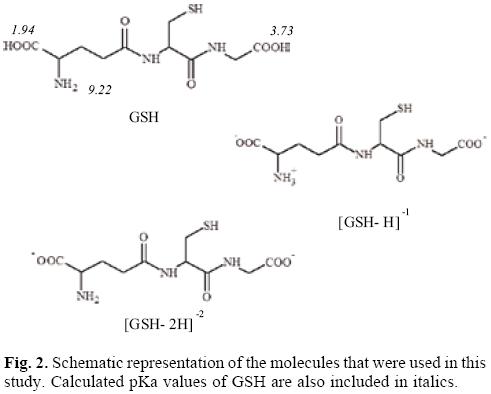
With the aim of study the electron donor-acceptor properties of these compounds, it is necessary to obtain the electronegativity for the donation (χ-) and for the electron acceptance (χ+). For this purpose, the cation and the anion were calculated with the optimized structures of the neutral molecule. The results for χ- and χ+ are shown in Table 2. Expectedly, as a result of the positive charge, these values are very high, indicating that copper complexes are bad electron donors and good electron acceptors. The electron donor-acceptor capacity of these compounds is compared with the electron donor-acceptor properties of other molecules which might be important in terms of cancer disease. These are the DNA nitrogen bases, the DNA nitrogen base pairs, certain free radicals which are responsible for oxidative stress and GSH which is apparently important as it is involved in the genotoxic activity of copper compounds [52-58]. The results are shown in Table 2. The optimized structure and the calculated [69] pKa values of GSH are shown in Figure 2. Under physiological conditions, a de-protonated species can be expected and for this reason in this study, we used (GSH-2H)-2 and (GSH-H)-1 (both represent two types of reduced glutathione).
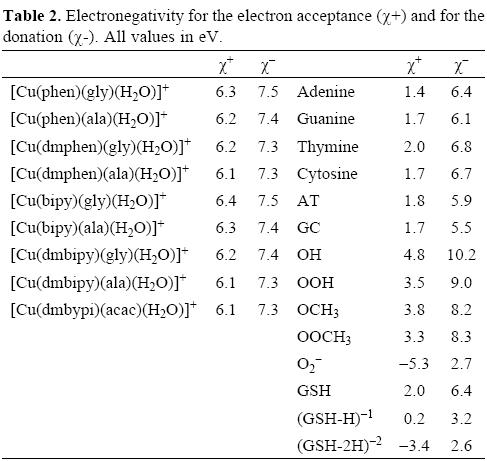
In order to facilitate the comparison, it is possible to construct a graph as we shown in Figure 3. In this Figure, all the molecules located in the upper right corner represent good electron acceptors and bad electron donors (χ- and χ+ have high values). These will thus remove electrons from the molecules placed in the lower left corner which represent good electron donors and bad electron acceptors (χ- and χ+ have low values). As is evident in Table 2 and Figure 3, χ- values for the nitrogen bases are lower than those for copper compounds. The greatest x- values are those pertaining to the neutral free radicals, indicating that these are the worst electron donors, whereas the superoxide radical anion (O2-•), (GHS-H)-1 and (GSH-2H)-2 represent good electron donors. This is a logical finding, as these molecules are negatively charged. Besides this, these results concur with the experimental information, because reduced glutathione molecule is considered to constitute an electron-rich antioxidant with significant reducing power.
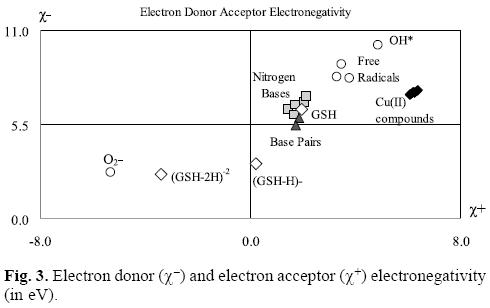
With respect to the electronegativity associated with acceptance, (χ+) copper compounds present the highest values thus making them the best electron acceptors. Apparently, copper compounds should be able to accept electrons from nitrogen bases, nitrogen base pairs, O2-•, (GSH-H)-1 and (GHS-2H)-2. They are not effective free radical scavengers of the neutral free radicals that we report in Table 2 and Figure 3, as copper compounds are not located at the top right of the neutral free radicals. Free radicals are worse electron donors than copper compounds, and not very good electron acceptors. Consequently, no charge transfer process will take place between neutral free radicals and copper compounds.
Analyzing the values in Table 2 and Figure 3, it is possible to see that copper compounds should be able to accept electrons from the superoxide radical anion, (GSH-H)-1 and (GSH-2H)-2. The donor electronegativity of these three molecules is very similar, so a comparable charge transfer process can be expected to occur from these anions to the copper complexes. If this is the case, Casiopeinas® operate by two possible mechanisms which may either help to prevent or avoid cancer disease. As good electron acceptors they may act as antiradicals by accepting electrons from the superoxide radical anion. Similar results were found previously for carotenoids [21-23], which are very well known antioxidants. It is worth noting that the electron acceptor capacity of the copper compounds reported in this work is almost twice that of the carotenoids. Since the superoxide radical anion may cause disorders associated with oxidative stress and because it is also a major source of other highly reactive oxygen species (ROS), it is possible to state that the antiradical activity of Casiopeinas® lies in their capacity to prevent the formation of oxidant ROS. This result is very important in view of the fact that previous suggestions regarding the mechanism operating in the case of Casiopeinas® [52] is related to reactive oxygen species (ROS) generation as a result of copper reduction, contradicting the results that we report here. If Casiopeinas® are good antiradicals, they should be able to prevent oxidative damage to lipids, proteins and DNA at the starting point of the free radical production reactions, as we illustrated in Figure 4. As it is well known that oxidative damage plays a role in carcinogenesis, Casiopeinas® would thus be good substances for preventing the formation of cancer cells.
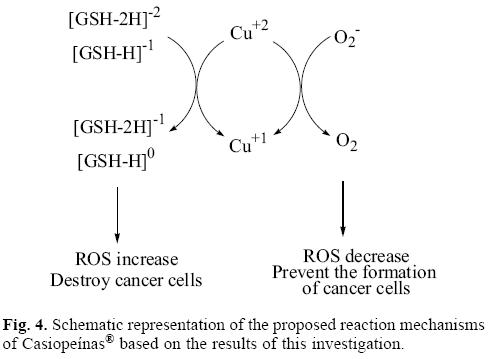
On the other hand, Casiopeinas® may also accept electrons from (GSH-H)-1 and (GSH-2H)-2. These results explain the dramatic drop in intracellular levels of GSH in human lung cancer cells, induced by the presence of one of these copper complexes, as shown in experiments [57]. The reduction of GSH by this redox process produces an increment in free radicals, which is also apparent in the experiment. It has been previously reported that oxidative stress is an important mechanism operating in a number of anticancer drugs. Apparently cancer cells exist in an elevated oxidative state, and therefore they are more vulnerable to the increment of the concentration of reactive oxygen species (ROS). The over-production of free radicals that Casiopeinas® apparently generate, leads to mitochondrial dysfunction and cell death because of the increment in oxidative stress. Cancer cells die faster than normal cells as a consequence of the oxidative stress, because they are more vulnerable.
Up to now, two different mechanisms can be considered to operate in the anticancer activity of Casiopeinas®. These copper complexes may be antiradical molecules which avoid the production of free radicals and as a consequence the generation of cancer cells, resulting from oxidative stress. The second mechanism operating here is thought to result from these molecules reducing the amount of (GSH-H)-1 and (GSH-2H)-2. As a result, they over-produce free radicals increasing oxidative stress, which kills cancer cells. GSH depletion can trigger suicide of the cell by a process known as apoptosis. In order to define which of these two mechanisms is the best, or under which conditions one is preferable to the other is undoubtedly an area requiring investigation.
Concluding remarks
Electronegativity for electron acceptance (χ+) and for donation (χ-) provides useful parameters for studying the electron-donor acceptor properties of molecules. Applying these parameters, it is possible to analyze the mechanism which is operating in anticancer agents of compounds such as Casiopeinas®.
Casiopeinas® are copper compounds which have been described previously. They represent good electron acceptors and are able to interact with good electron donors such as superoxide (O2-•) radical and reduced glutathione [(GSH-H)1- and GSH-2H)2-]. These copper compounds manifested the highest values in terms of accepting electronegativity (%+) with respect to O2-•, neutral free radicals, nitrogen bases, nitrogen base pairs and glutathione, ie. Casiopeinas® are the best electron acceptors out of all these compounds.
The oxidative damage in the cell plays an important role in carcinogenesis. When Casiopeinas® interact with O2-• they are able to preclude cancer because copper complexes accept electrons from O2-• preventing oxidative damage from occurring in the cell. On the other hand, when Casiopeinas® interact with (GSH-H)1- and (GSH-2H)2- they can inhibit cancer cells as GSH depletion is able to trigger cell suicide by a process known as apoptosis. The reaction mechanism of Casiopeinas® is not well known, but from these results we suggest two probable mechanisms derived from these copper compounds, for the treatment of cancer.
Computational Details
Density functional approximation [59-61] as implemented in Gaussian 09 [62] was used for all calculations. Full geometry optimizations without symmetry constraints and frequency analysis were carried out for all the stationary points using the three parameters B3LYP [63-65] density functional and 6-31+G(d,p) basis sets. Harmonic frequency analyses made it possible for us to verify optimized minima. Local minima were identified by the number of imaginary frequencies (NIMAG = 0). In order to compute I and A, further single-point calculations are necessary. I is calculated as the difference between the energy of the cation and the neutral molecule, assuming that both of these have the ground-state nuclear configuration of the neutral molecule. A is also calculated as vertical, and represents the energy difference between the neutral and the anion, calculated with the ground-state nuclear configuration of the neutral molecule.
Previous studies indicate that DFT reproduces equilibrium geometries and relative stabilities with hybrid function-als, which partially include the Hartree-Fock exchange energy. These results are largely consistent with those obtained using the Moller-Plesset perturbational theory at second order and basis sets of medium quality, such as 6-31G(d,p), and cc-pVDZ [66, 67].
The geometrical optimization of (GSH-H)- was not possible in gas phase, since the H atom that was bound to the N atom in the initial geometry binds to the oxygen during optimization. In order to have the molecule with NH3+ and COO-, which is the formula expected under physiological conditions, optimization was achieved in water, using the SMD option of Gaussian09. The optimized structure in water was used to calculate the energy (without optimization) of this system, as neutral, anionic and cationic in gas phase. The pKa values were obtained using the Marvin Sketch code from Chem Axon.
Acknowledgments
This study was made possible due to funding from the Consejo Nacional de Ciencia y Tecnología (CONACyT), as well as resources provided by the Instituto de Investigaciones en Materiales IIM, UNAM. The work was carried out, using a KanBalam supercomputer, provided by DGTIC, UNAM. The authors would like to thank Elizabeth Hernández-Marin for helpful discussions, and acknowledge both Oralia L. Jiménez A and Maria Teresa Vázquez for their technical support. MA is grateful for scholarship from CONACYT-México.
References
1. Parr, R. G.; Yang, W. Density-Functional Theory of Atoms and Molecules; Oxford University Press: New York, 1989. [ Links ]
2. Dreizler, R. M.; Gross, E. K. U. Density Functional Theory; Springer: Berlin, 1990. [ Links ]
3. Perdew, J. P.; Kurth, S. In A Primer in Density Functional Theory, Fiolhais, C.; Nogueira, F.; Marques, M. A. L. Eds.; Springer: Berlin, 2003; p. 1. [ Links ]
4. Koch, W.; Holthausen, M. C. A Chemist's Guide to Density Functional Theory; Wiley-VCH: New York, 2000. [ Links ]
5. Pearson, R. G. Chemical Hardness: Applications from Molecules to Solids; Wiley-VCH: Oxford, 1997. [ Links ]
6. Parr, R. G.; Donnelly, R. A.; Levy, M.; Palke, W. E. J. Chem. Phys. 1978, 68, 3801-3807. [ Links ]
7. Parr, R. G.; Pearson, R. G. J. Am. Chem. Soc. 1983, 705, 75127516. [ Links ]
8. Gázquez, J. L. In Chemical Hardness, Sen, K. D.; Mingos, D. M. P. Eds.; Springer-Verlag: Berlin, 1993; pp 27-43. [ Links ]
9. Gázquez, J. L. J. Phys. Chem. A 1997, 101, 4657-4659. [ Links ]
10. Gázquez, J. L.; Martinez, A.; Méndez, F. J. Phys. Chem. 1993, 97, 4059-4063. [ Links ]
11. Gázquez, J. L. J. Phys. Chem. A 1997, 101, 9464-9469. [ Links ]
12. Gázquez, J. L.; Méndez, F. J. Phys. Chem. 1994, 98, 4591-4593. [ Links ]
13. Méndez, F.; Gázquez, J. L. Proceedings of the Indian Academy of Sciences-Chemical Sciences 1994, 106, 183-193. [ Links ]
14. Méndez, F.; Gázquez, J. L. J. Am. Chem. Soc. 1994, 116, 9298-9301. [ Links ]
15. Gázquez, J.L.; Cedillo, A.; Vela, A. J. Phys. Chem. A. 2007, 111, 1966.1970 [ Links ]
16. Gázquez. J. L. J. Mex. Chem. Soc. 2008, 52, 3-10 [ Links ]
17. Ramirez-Ramirez, J. Z.; Vargas, R.; Garza, J.; Gázquez, J. L. J. Phys. Chem. A. 2010, 114, 7945-7951. [ Links ]
18. Martinez, A.; Rodriguez-Gironés, M. A.; Barbosa, A. ; Costas, M. J. Phys. Chem. A 2008, 112, 9037-9042. [ Links ]
19. Martinez, A. J. Phys. Chem. B 2009, 113, 3212-3217. [ Links ]
20. Martinez, A. J. Phys. Chem. B 2009, 113, 4915-4921. [ Links ]
21. Martinez, A.; Vargas, R.; Galano, A. J. Phys. Chem. B 2009, 113, 12113-12120. [ Links ]
22. Martinez, A.; Rodriguez-Gironés, M. A.; Barbosa, A. Ardeola 2009, 56, 287-294 [ Links ]
23. Galano, A.; Vargas, R.; Martinez, A. Phys. Chem. Chem. Phys. 2010, 12, 193-200. [ Links ]
24. Martinez, A.; Barbosa, A. Theor. Chem. Acc. 2010, 127, 485-492. [ Links ]
25. Martinez, A.; Vargas, R. New J. of Chem. 2010, 34, 2988-2995. [ Links ]
26. Martinez, A. J. Phys. Chem. C 2010, 114, 21240-21246. [ Links ]
27. Burton, G.W.; Ingold, K.U. Science, 1984, 224, 569-573. [ Links ]
28. Edge, R.; McGarvey, D.J.; Truscott, T.G. J. Photochemistry and Photobiology B: Biology. 1997, 41, 189-200. [ Links ]
29. Edge, R.; Land, E.J. McGarvey, D.J.; Mulroy,L.; Truscott, T.G. J. Am. Chem. Soc. 1998, 120, 4087-4090. [ Links ]
30. Galano, A. J. Phys. Chem. B. 2007, 111, 12898-12908. [ Links ]
31. Martin, H.D.; Jäger, C.; Ruck, C.; Schmidt, M.; Walsh, R., Paust, J. J. Prakt. Chem. 1999, 341, 302-308. [ Links ]
32. Mairanosvky, V.G.; Engovatov, A.A.; Ioffe, N.T.; Samokhvalov, G. I. J. Electroanal.Chem. 1975, 66, 123-137. [ Links ]
33. Polyakov, N.E.; Leshina, T.A.; Konovalova, T.A.; Kispert, L.D. Free Radical Biology & Medicine, 2001, 31, 398-404. [ Links ]
34. Polyakov, N.E.; Kruppa, A.I.; Leshina, T.V.; Konovalova, T.A.; Kispert,L.D. Free Radical Biology & Medicine, 2001, 31, 43-52. [ Links ]
35. Mortensen, A.; Skibsted, L.H.; Sampson, J.; Rice-Evans, C.; Everett, S.A. FEBS Letters, 1997, 418, 91-97. [ Links ]
36. Jeevarajan, A.S.; Khaleb, M.; Kispert, L.D. J. Phys. Chem . 1994, 98, 7777-7781. [ Links ]
37. Halliwell, B. Free Rad. Res. 1996, 25, 439-454. [ Links ]
38. Prior, R.L.; Cao, G. Free Radical Biology & Medicine. 1999, 27, 1173-1181. [ Links ]
39. Mortensen, A.; Skibsted, L.H. J. Agric. Food Chem. 1997, 45, 2970-2977. [ Links ]
40. Baskin, S.I.; Salem, H. Oxidants, Antioxidants and the Free Radicals. Taylor & Francis Ed. 1997, Washington. [ Links ]
41. Woodall, A.A.; Britton, G.; Jackson, M.J. Biochimica et Biophysica Acta, 1997, 1336, 575-586. [ Links ]
42. Woodall, A. A.; Lee, WMS.; Weesie, R.J.; Jackson,M.J.; Britton, G. Biochimica et Biophysica Acta -General Subjects, 1997, 1336, 33-42. [ Links ]
43. Guo, J.D.; Luo, Y.; Himo, F. Chem. Phys. Lett. 2002, 366, 73-81. [ Links ]
44. Burr, M. L. J. of Human Nutrition and Dietetics 1994, 7, 409-416. [ Links ]
45. Ritenbaugh, C. Nutrition Today 1987, 21, 14-19. [ Links ]
46. Connett, J. E.; Kuller, L. H.; Kjelsberg, M. O.; Polk, B. F.; Collins, G.; Rider, A.; Hulley, S. B.; Cancer, 1989, 64, 126-134. [ Links ]
47. Sata, J. A.; Littman, A.; Slatore, C. G.; Galanko, J. A.; White, E. Am. J. of Epidemiology 2009, 169, 815-828. [ Links ]
48. Lambert, J. D.; Elias, R. J. Review Article Archives of Biochemistry and Biophysics, 2010, 501, 65-72. [ Links ]
49. Solans, X.; Ruiz-Ramirez, L.; Martinez, A.; Gasque, L.;Briansó, J.L. Acta Cryst. 1988, C44, 628-631. [ Links ]
50. Solans, X.; Ruiz-Ramirez, L.; Martinez, A.; Gasque, L.; Moreno-Esparza, R. Acta Cryst. 1992, C48, 1785-1788. [ Links ]
51. Solans, X.; Ruiz-Ramirez, L.; Martinez, A.; Gasque, L.; Moreno-Esparza, R. Acta Cryst. 1993, C49, 890-893. [ Links ]
52. Serment-Guerrero, J.; Cano-Sánchez, P.; Reyes-Perez, E.; Velazquez-Garcia,F.; Bravo-Gómez,M.E.; Ruiz-Azura, L. Toxicology in Vitro, 2011, 25, 1376-1384. [ Links ]
53. Ruiz-Ramirez, L.; de la Rosa, M.E.; Gracia-Mora, I.; Mendoza, G. ; Pérez, G.; Ferrer-Sueta, G.;Tovar, A.; Breña, M.; Gutiérrez, P.; Cruces Martinez, M.P.; Pimentel, E.; Natarajan, A. T. J. Inorg. Biochem. 1995, 59, 207. [ Links ]
54. Ruiz-Ramirez, L.; Gracia-Mora, I.; de la Rosa, M.E.; Sumano, H. ; Gómez, C.; Arenas, F.; Gómez, E.; Pimentel, E.; Cruces, M. J. Inorg. Biochem. 1993, 51, 406. [ Links ]
55. Tovar-Tovar, A.; Ruiz-Ramirez, L.; Moreno-Esparza,R.; Briansó, J.L. J. Inorg. Biochem. 1995, 59, 206. [ Links ]
56. De Vizacaya-Ruiz,A.; Rivero-Muller, A.; Ruiz-Ramirez, L.; Kass, G.E.N.; Kelland, L.R.; Orr, R.M.; Dobrota, M. Toxicology in Vitro, 2000, 14, 1-5 [ Links ]
57. Kachadourian, R.; Brechbuhl, H.M.; Ruiz-Azuara, L.; Gracia-Mora, I.; Day, B.J. Toxicology 2010, 268, 176-183. [ Links ]
58. Alemón-Medina, R.;Breña-Valle, M.; Muñoz-Sánchez, J. L.; Gracia-Mora, I.; Ruiz-Azuara, L. Cancer Chemotherapy and Pharmacology 2007, 60, 219-228. [ Links ]
59. Kohn, W.; Becke, A. D.; Parr, R. G. J. Phys. Chem. 1996, 100, 12974-12980. [ Links ]
60. Hohenberg, P.; Kohn, W. Phys. Rev. 1964, 136, B864-B871. [ Links ]
61. Kohn, W.; Sham, L. J. Phys. Rev. 1965, 140, A1133-A1138. [ Links ]
62. Gaussian 09, Revision A.08, Frisch, M. J., Trucks, G. W., Schlegel, H. B., Scuseria, G. E., Robb, M. A., Cheeseman, J. R., Scalmani, G., Barone, V., Mennucci, B., Petersson, G. A., Nakatsuji, H., Caricato, M., Li, X., Hratchian, H. P., Izmaylov, A. F., Bloino, J., Zheng, G., Sonnenberg, J. L., Hada, M., Ehara, M., Toyota, K., Fukuda, R., Hasegawa, J., Ishida, M., Nakajima, T., Honda, Y., Kitao, O., Nakai, H., Vreven, T., Montgomery, Jr., J. A., Peralta, J. E., Ogliaro, F., Bearpark, M., Heyd, J. J., Brothers, E., Kudin, K. N., Staroverov, V. N., Kobayashi, R., Normand, J., Raghavachari, K., Rendell, A., Burant, J. C. Iyengar, S. S. Tomasi, J. Cossi, M. Rega, Millam, N. J., Klene, M. Knox, J. E., Cross, J. B., Bakken, V., Adamo, C., Jaramillo, J., Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, R. Martin, R. L., Morokuma, K., Zakrzewski, V. G., Voth, G. A., Salvador, P., Dannenberg, J. J., Dapprich, S., Daniels, A. D., Farkas, O., Foresman, J. B., Ortiz, J. V., Cioslowski, J., and Fox, D. J., Gaussian, Inc., Wallingford CT, 2009. [ Links ]
63. Becke, A. D. Phys. Rev. A 1988, 38, 3098-3100. [ Links ]
64. Mielich, B.; Savin, A.; Stoll, H.; Peuss, H. Chem. Phys. Lett. 1989, 157, 200-206. [ Links ]
65. Lee, C.; Yang, W.; Parr, R. G. Phys. Rev. B 1988, 37, 785-789. [ Links ]
66. Maller, C.; Plesset, M. S. Phys. Rev. 1934, 46, 618-622. [ Links ]
67. Saebo, S.; Almlof, J. Chem. Phys. Lett. 1989, 154, 83-89. [ Links ]
68. Cioslowski, J. J. Am. Chem. Soc., 1989, 111, 8333-36. [ Links ]
69. Marin Sketch, version 5.5.0. Chem Axon Ltd. [ Links ]
* The asterisk indicates the name of the author to whom inquires about the paper should be addressed.
Dedicated to our friend and teacher for many years José Luis Gázquez.

