










Servicios Personalizados
Revista
Articulo
 Inglés (pdf)
Inglés (pdf)
 Artículo en XML
Artículo en XML Referencias del artículo
Referencias del artículo
 Enviar artículo por email
Enviar artículo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO -
 Accesos
Accesos
Links relacionados
-
 Similares en
SciELO
Similares en
SciELO
Compartir

 Permalink
PermalinkJournal of the Mexican Chemical Society
versión impresa ISSN 1870-249X
J. Mex. Chem. Soc vol.56 no.3 Ciudad de México jul./sep. 2012
Artículo
Role of Lithium Decoration on Hydrogen Storage Potential
Sudip Pan, Swastika Banerjee, and Pratim K. Chattaraj*
Department of Chemistry and Center for Theoretical Studies, Indian Institute of Technology, Kharagpur-721303, India. pkc@chem.iitkgp.ernet.in
Received August 02, 2011.
Accepted February 20, 2012.
Abstract
Hydrogen storage potential of two sets of lithium containing systems, viz., Li-doped borazine derivatives and various bond-stretch isomers of Li3Al4- is studied at the B3LYP/6-311+G(d) level of theory occasionally supplemented by the results from the associated MP2/6-31+G(d) calculations. Negative values of interaction energy, reaction enthalpy, reaction electrophilicity, and desorption energies for the gradual hydrogen-trapping processes justify the efficacy of these systems as the hydrogen storage material. Presence of Li as well as aromaticity improves the situation. Various conceptual density functional theory based reactivity descriptors like electronegativity, hardness, and electrophilicity and the associated electronic structure principles such as the principles of maximum hardness and minimum electrophilicity lend additional support.
Key words: Hydrogen Storage, Conceptual DFT, Hardness, Electro-philicity, NICS, Aromaticity.
Resumen
En este artículo se estudia el potencial para el almacenamiento de hidrógeno de dos conjuntos de sistemas que contienen litio, del tipo de derivados de borazina dopados con Li y varios isómeros de Li3Al4-, al nivel de teoría B3LYP/6-311+G(d), complementándose ocasionalmente por resultados de otros cálculos al nivel MP2/6-31+G(d). Se obtienen valores negativos para la energía de interacción, entalpia de reacción, electrofilicidad de reacción, y energía de desorción para todos los procesos de atrapamiento gradual de hidrógeno, justificándose así la eficacia de estos sistemas como materiales almacenadores de hidrógeno. La presencia de Li así como la aromaticidad mejoran la situación. Varios descriptores de reactividad basados en la teoría de funcionales de la densidad conceptual, como la electronegatividad, dureza y electrofilicidad, así como los principios asociados de estructura electrónica —tales como el principio de máxima dureza y el de mínima electrofilicidad— dan apoyo adicional a las conclusiones.
Palabras clave: Almacenamiento de hidrógeno, DFT conceptual, dureza, electrofilicidad, NICS, aromaticidad.
Introduction
Hydrogen, the third most abundant element on earth has been considered to be an ideal alternative energy carrier for its renewability and non-polluting nature [1, 2]. Unlike petroleum, hydrogen can be easily generated from renewable energy resources which further eliminate the production of oxides of nitrogen and sulfur, green-house gases like carbon dioxide and methane as by-products thereby eradicating further scopes of environmental pollution. The criteria of an ideal hydrogen storage material are (i) high volumetric and gravimetric densities, (ii) fast kinetics for adsorption and desorption at ambient conditions, (iii) favorable enthalpies and dissociative chemisorption energy for hydrogen adsorption and desorption, (iv) recyclability and (v) cost effectiveness of material [3, 4]. To achieve reversible hydrogen adsorption and desorption at near ambient condition, the hydrogen binding energy should be somewhat intermediate between that of physisorption and chemisorption [5]. But the storage of gaseous hydrogen in a practical sense creates difficulties as the materials that can trap hydrogen do not meet all the requirements to achieve the targets of a gravimetric density of 6.0% in 2010 and 9.0% in 2015 and volumetric capacity of 45 gL-1 in 2010 and 81gL-1 in 2015 set by the Department of Energy (DOE) [6]. Therefore searching new material for hydrogen storage is an important and active area of research.
In recent years hydrogen adsorption has been tested in large number of materials such as carbon, boron based nano-materials [7-12], alanates [13, 14], clathrates [15-18], borates [19], zeolites [20], metalhydrides [21, 22], metal-organic frameworks (MOFs) [23-25], covalent-organic frameworks (COFs) [26, 27], zeolitic-imidazole frameworks (ZIFs) [28-30], porous silica etc. [31]. Though the MOFs and COFs are having high surface area, the hydrogen binding energy is very poor.
The hydrogen binding energy can be improved by decorating organic linker with metal atoms / ions like lithium, where the metal atom gets partially charged and binds molecular hydrogen strongly [32-35]. In this work an attempt has been made to study the binding of hydrogen with a host of lithium substituted borazine derivative (B3N3F3Li3) and its different Li+ doped systems (B3N3F3Li4+, B3N3F3Li52+). A sandwich type complex, [(B3N3F3Li4)2F]+ can be formed by bridging two B3N3F3Li4+ systems via an F- and that has also been found to be a potential hydrogen trapping substance. Here an investigation has also been made to examine the hydrogen storage capacity of different geometrical isomers of Li3Al4- clusters. In this regard, seven pairs of possible bond-stretch isomers of Li3Al4- clusters [36] have been analyzed.
The occurrence of bond-stretch isomerism is now experimentally established via X-ray crystal structure determination. These are also called distortion isomers. The term "distortional isomerism" was first proposed by Chatt and Manojlovic-Muir in 1971 to characterize metallic complexes that differ only by the length of one or several bonds. Later they have been termed as bond-stretch isomers [37-40]. In Li3Al4- σ-aromaticity component predominates over π-(anti)aromaticity component making the system overall aromatic in nature. These cluster molecules are of "fleeting" type [41] and at most kinetically stable.
Moreover, there exists a possibility [42] of ''bond-stretch isomerism'' in these systems depending on which particular local minima (and/or the global minimum) it is stuck in. In this work, the bonding, aromaticity, and various isomeric local minimum structures of Li3Al4- cluster and the possibility of bond-stretch isomerism and their associated aromaticity have been studied.
Structure, stability and reactivity of these systems with or without H2 trapping have been studied using conceptual density functional theory (CDFT) [43-46] based reactivity descriptors like electronegativity [47, 48] (χ), hardness [49-51] (η), electro-philicity [52-54] (ω) and the local variants like atomic charges [55] (Qk). The stability of the resulting hydrogen-bound complexes may be understood from the corresponding interaction energies (ΔE) and reaction electrophilicities (Δω) of plausible trapping reactions. The exothermicity of these given reactions towards formation of the stable H2-trapped analogues can be justified from the negative reaction enthalpy (ΔH) values. Further, for most of the hydrogen bound clusters, hydrogen prefers mostly to coordinate with the alkali metal center (Li) in its molecular (dihydrogen) form. Molecular hydrogen does not have a dipole moment, but it has a strong quadrupole moment and polarizability. Hence, a charged site can bind with the molecular hydrogen through the dipole-quadrupole and dipole-induced dipole types of interactions.
Theoretical Background
In general, thermodynamic stability of a molecular system can be meaningfully predicted by the establishment of some associated molecular electronic structure principles like the Maximum Hardness Principle [56-58] (MHP) together with the Minimum Polarizability Principle [59, 60] (MPP) and Minimum Electrophilicity Principle [61, 62] (MEP).
For an N-electron system having total energy E, the electronegativity [47, 48] (χ) and hardness [49-51] (η) can be defined as follow
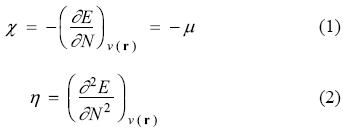
where v(r) and m are external and chemical potentials respectively. Electrophilicity [52-54] (ω) is defined as

Applying the finite difference approximations to equations 1 and 2, χ and η can be expressed as

and

where I and A are the ionization potential and electron affinity of the system, respectively. Further, according to the Koop-mans' theorem [63] the ionization potential (I) and electron affinity (A) of a molecular system can be expressed in terms of the energies of the frontier molecular orbitals (FMOs) as:
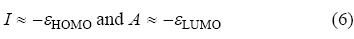
It may be noted that Koopmans' theorem [63] is strictly valid within the Hartree-Fock theory. In Kohn-Sham calculations one may use Janak's theorem [64].
According to ΔSCF method, the ionization potential (I) and electron affinity (A) of the system respectively are computed in terms of the energies of the N and N ± 1 electron systems. For an N-electron system with energy E(N), they may be expressed as follows:
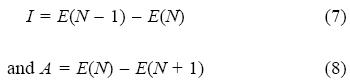
Computational Details
The geometry optimization and subsequent frequency calculation of all borazine derivatives and their corresponding hydrogen-trapped analogues have been carried out at the B3LYP level of theory using 6-311+G(d) basis set whereas in case of all bond stretch isomers of Li3Al4-, both B3LYP/6-311+G(d) and MP2/6-31+G(d) levels of calculations have been performed with the aid of the GAUSSIAN 03 program package [65]. For the hydrogen trapped analogues, at first free optimizations followed by frequency calculations have been carried out. There very often exist several soft modes of imaginary frequencies for the hydrogen trapped case. Then the minima (NIMAG = 0) on the potential surface have been attained by following those modes corresponding to imaginary frequencies. There also exist several possible local minimum structures for these species. The vertical electron detachment energies of Li3Al4- isomers have been carried out by using Koopmans' theorem [63] at HF level and ASCF technique at MP2, B3LYP and CCSD levels of theory. Outer valence Green's functional has also been used to determine vertical electron detachment energy. The ionization potential (I) and electron affinity (A) values have been calculated using ΔSCF technique at the MP2 level whereas Koopmans' theorem [63] has been followed in case of B3LYP calculation. A Mulliken population analysis (MPA) scheme has been adopted to calculate the atomic charges (Qk) [55]. The NICS(0) and NICS(1) [66] values have been calculated. The frontier molecular orbital pictures have been obtained through the GAUSSVIEW 03 package [65].
To calculate interaction energy per H2 molecule and desorption energy per H2 molecule the following expressions have been used.
Interaction energy/H2 molecule,

Desorption energy,

where S is the hydrogen-trapped systems.
A different definition of ΔEDE in terms of the desorption of H atoms has been used elsewhere [67].
Results and Discussion
Li-doped Borazine Derivatives
The optimized structures at B3LYP level of theory using 6-311+G(d) basis set of the considered systems B3N3F3Li3, B3N3F3Li4+, B3N3F3Li52+ and [(B3N3F3Li4)2F]+ and their some representative H2 trapped analogues have been given in Figure 1. These structures correspond to minima on the potential energy surface at the used level of theory. The interaction energy (ΔE), change in free energy (ΔG) and enthalpy (ΔH) of Li+ doping processes on B3N3F3Li3 and bridging of B3N3F3Li4+ by an F- have been given in Table S1 (as supporting information). Negative interaction energies show the stability of the complexes towards dissociation which is needed for a recyclable hydrogen storage material whereas spontaneity and exothermicity of the doping and bridging processes are indicated by large negative ΔG and ΔH values. Interaction energy per hydrogen molecule (ΔE), reaction enthalpy (ΔH), desorption energy per hydrogen molecule (ΔEDE), and conceptual DFT based global reactivity descriptors like electronegativity (χ), hardness (η) and electrophilicity (ω) of the bare as well as several H2 bound B3N3F3Li3, B3N3F3Li4+, B3N3F3Li52+ and [(B3N3F3Li4)2F]+ systems have been shown in Table 1. A careful scrutiny of Table 1 reveals that the electronegativity of the different nH2 bound B3N3F3Li3 and B3N3F3Li4+ systems show a decreasing trend while the chemical hardnesses and electrophilicities are on an average of increasing trend and decreasing trend respectively whereas in case of nH2 bound B3N3F3Li52+ system, hardness has increased upto 10H2 and then it has decreased whereas electrophilicity has decreased upto 10H2 then it has slightly increased for 14H2 storing indicating greater stability of 10H2@ B3N3F3Li52+ over 14H2@B3N3F3Li52+. In case of nH2 bound [(B3N3F3Li4)2F]+, it has also been found that hardness has increased for 6H2 and then it has very slightly decreased for 12H2 and 17H2 binding but electrophilicity has a decreasing trend with gradual cluster growth (upon H2 binding). Again for B3N3F3Li3 system, it has been found that the interaction energy per hydrogen molecule is quite low. First three hydrogen molecules have been adsorbed with an interaction energy of -1.8 kcal/mole per H2 molecule, whereas for 6H2 and 9H2 trapped analogues, the hydrogen molecules have been found to get adsorbed with an interaction energy of -1.4 and -1.1 kcal/mole per H2 molecule respectively. Again for this system, reaction enthalpies for the several hydrogen binding processes are quite less negative and finally become positive for adsorption of the last three hydrogen molecules. Upon Li+ doping on B3N3F3Li3 system, it has been found that interaction energies and reaction enthalpies improve significantly. In the case of B3N3F3Li4+, the first four hydrogen molecules have been found to get adsorbed with an interaction energy of -2.6 kcal/mol per H2 molecule, whereas for 8H2 and 12H2 bound analogues, the interaction energies have been found to be -2.1 and -1.6 kcal/mol per H2 molecule respectively. The reaction enthalpies are also quite negative indicating the exothermicity of H2 trapping process. In the case of B3N3F3Li52+, the first five H2 molecules have been found to get adsorbed with a reasonably good interaction energy of -4.1 kcal/mol per H2 molecule, whereas for 10H2 and 14H2 trapped analogues, the interaction energies have been found to be -3.3 and -2.8 kcal/mol per H2 molecule respectively. The reaction enthalpies (ΔH) are also quite highly negative. In the case of [(B3N3F3Li4)2F]+ system, the first six hydrogen molecules have been adsorbed with an interaction energy of -2.4 kcal/mol per H2 molecule whereas the interaction energies for 12H2 and 17H2 bound analogues are -1.9 and -1.5 kcal/mol per H2 molecule respectively and from the reaction enthalpy (ΔH) values for this system it can be told that the H2 trapping processes of this system are quite exothermic. The desorption energies per H2 molecule (ΔEDE) of all the systems are also in the favorable range which is needed for a system to be a good recyclable hydrogen storage material. In Table 1, the desorption energies per H2 molecule (ΔEDE) correspond to the corresponding quantities per H2 molecule for the simultaneous desorption of m molecules where m = 3 for B3N3F3Li3, m = 4 for B3N3F3Li4+, m = 5, 4 for B3N3F3Li52+ and m = 6, 5 for [(B3N3F3Li4)2F]+ cases. The values of NICS(0) and NICS(1) for all considered borazine derivatives and their corresponding hydrogen trapped analogues have been given in Table S2 (supporting information). From the NICS values, it has also been found that B3N3F3Li3, B3N3F3Li4+ and both rings of [(B3N3F3Li4)2F]+ are almost non-aromatic or very slightly aromatic whereas B3N3F3Li52+ gets some considerable degree of aromaticity. It has also been shown that for all the systems the variations of both NICS(0) and NICS(1) are quite small with gradual H2 trapping. The important frontier molecular orbitals (HOMO and LUMO) of the considered systems and their H2 trapped analogues have been given in Figure S9 (supporting information). In the case of B3N3F3Li3, B3N3F3Li4+, B3N3F3Li52+ and [(B3N3F3Li4)2F]+, the gravimetric densities of hydrogen have been found to be 10.65, 13.19, 14.52 and 9.22 wt% respectively. It may be relevant to mention here that appropriate counter ions are needed to tackle the Coulomb instability of these charged clusters which would somewhat reduce the corresponding gravimetric values of hydrogen adsorbed.
Li3Al4- Isomers
Here seven geometrical isomers of Li3Al4- namely Fork, Hood, Cs , C1, Rabbit C2v, Rabbit Cs, Scooter C1 and their singlet as well as triplet state geometries (seven bond-stretch isomer-ic pairs) have also been studied. Figure 2 depicts the optimized geometries and the frontier molecular orbital pictures of Li3Al4- isomers and their corresponding highest hydrogen trapped analogues. NICS(0) values of the isomeric clusters and their corresponding highest hydrogen-trapped analogues have also been provided in Figure 2. The NICS(0) values reveal that triplet analogues are strongly aromatic whereas the singlet components are reasonably aromatic in the respective bond-stretch isomeric pairs. Again an increase of aromaticity upon H2 trapping also dictates the stability of the hydrogen-bound systems with respect to the unbound systems. There is a reversal of the molecular electronic configurations viz., in one case the HOMO is of π-type and the LUMO is of σ-type which gets reversed for the other isomer. Frontier orbitals for pair of bond stretch isomers corresponding to Rabbit isomers (C2v and Cs) and C1 and Cs isomers remain same. The HOMO and LUMO pictures of the systems show the same symmetry in both H2-bound and free Li3Al4- cluster, as expected when the hydrogens retain their molecular nature. In these systems, trapping of the hydrogen in molecular form is in between phy-sisorption and chemisorption which is the requirement for a good hydrogen storage material. The optimized geometries of the hydrogen trapped analogues of scooter Li3Al4- isomer have been given in Figure S1. The vertical electron detachment energies of Li3Al4- isomers at various levels of theory have been provided in Table 2. Positive values of vertical electron detachment energies of Li3Al4- isomers at all the studied levels of theory imply its gas phase stability with respect to spontaneous emission of electron. Total energies, electronegativity, hardness and electrophilicity of Li3Al4- bond-stretch isomeric pairs (of singlet and triplet spin multiplicities), calculated at B3LYP and MP2 levels of theory have been reported in Table S3 (supporting information) and Table 3 respectively. A comparison of the potency of the various bond-stretch isomeric (Fork, Hood, Cs, C1, Rabbit C2v, Rabbit Cs, Scooter C1) Li3Al4- clusters (in both singlet and triplet states) towards binding molecular hydrogen has been made at two different (B3LYP and MP2) levels of theory. The important global reactivity descriptors like electronegativity (χ), hardness (η) and electrophilicity (ω) and reaction electrophilicity (Δω) for all interacting isomeric clusters, have been provided in Tables S4a-S10a (supporting information). The negative χ values imply that these systems do not want to accept further electrons. In presence of counter-ions χ values become positive [68]. The n values have been found to correlate nicely with that of w as relevant from Tables S4a-S10a (supporting information) and the corresponding Figures S2-S8 (supporting information). The hardness (η) increases uniformly with a more or less decrease in the electrophilicity (ω) values thereby corroborating the associated principles of maximum hardness and minimum electrophilicity which justify further stability of the clusters upon hydrogen trapping. The interaction energy per hydrogen molecule (ΔE), reaction enthalpy (AH), and sequential desorption energy per H2 molecule (ΔEDE) calculated in B3LYP level of theory for all the isomeric clusters of Li3Al4- have been given in Tables S4b-S10b (supporting information) whereas the interaction energy per hydrogen molecule (ΔE) and sequential desorption energy per H2 molecule (ΔEDE) calculated in MP2 level of theory have been provided in Tables 4 and 5 for Rabbit Cs and C1 isomeric clusters respectively. A scrutiny of Tables 4 and 5 shows that all the interaction energy terms are negative and the sequential desorption energies per H2 molecule (all positive) are also in the favorable range. Here it should be mentioned that in the case of Tables 4 and 5, the desorption energies per H2 molecule have been calculated sequentially by using equation 10 with m = 1. In case of other isomeric clusters, the results obtained from the MP2 level have been provided in Tables S4c-S8c (supporting information). A quick look at Tables S4b-S10b (supporting information) and S4c-S8c (supporting information) for some plausible trapping reactions conceived between the Li3Al4- isomeric clusters and hydrogen molecules reveals that for all the reactions, the corresponding interaction energies (ΔE) and reaction electrophilicities (Δω) are negative. The situation becomes even more enthusing when the associated reaction enthalpy values also turn negative which eventually lends ample justification towards thermodynamic exothermicity for all the given trapping reactions and thereby rendering stability to the resultant H2-trapped complexes. The thermodynamic exothermicity accompanied by favorable interaction energy per hydrogen molecule for the given hydrogen binding reactions computed at the B3LYP level of theory therefore lends ample support towards the plausible usage of the Li3Al4- cluster as effective hydrogen storage material. Again the favorable reaction electrophilicity (Δω) values give support towards governing the feasibility of the given trapping reactions. The variation of Mulliken atomic charges on the Li centers of Rabbit-Cs with gradual hydrogen trapping has been provided in Table 6 and for the other isomeric clusters the same has been given in Tables S4d-S9d. The variations of charges of the Li centers upon hydrogen loading suggest that charge transfer takes place between Li centers and hydrogen molecule. Both cationic and anionic clusters need suitable counter-ions for stabilization vis-à-vis Coulomb instability [69].
Concluding Remarks
In this investigation, hydrogen storage capacity of a new Li substituted borazine derivative (B3N3F3Li3) has been explored. It has been found that interaction energy per hydrogen molecule and hydrogen adsorption enthalpy can be significantly improved by two successive Li+ doping. A new sandwich type of complex [(B3N3F3Li4)2F]+ has also been designed by bridging two B3N3F3Li4+ systems via an F- and it has also been found to be a good hydrogen storage substance. The variation of different global reactivity descriptors as a function of the gradual hydrogen loading suggests the stability of the hydrogen bound systems. Here an investigation has also been made to examine the hydrogen storage capacity of different geometrical isomers of Li3Al4- clusters. In this regard seven pairs of possible bond-stretch isomers (Li3Al4- clusters) in singlet and triplet states have been observed. Associated vertical detachment energies highlight their stability related to electron detachment. The nucleus independent chemical shift values suggest that all these systems are aromatic. In most cases for each pair of isomers, the frontier molecular orbital configurations are of opposite types. It may be looked into in the light of the test prescribed by Zubarev and Boldyrev [70]. The interaction energy per hydrogen molecule (ΔE), hardness (η) and electrophilicity (ω) of the poly-hydrogen bound Li3Al4- cluster suggest a gradual increment in stability upon trapping. The NICS (0) values for respective rings of the free as well as hydrogen-trapped systems are negative. Thus, the presence of an "all-metal aromaticity" in the different rings has been verified. These Li3Al4- isomeric clusters can be fruitfully applied as trapping materials for molecular hydrogen. One can remove the Coulomb instability and stabilize these ionic clusters in presence of some suitable counter ions and it is possible to make a 3D network by using suitable ionic linkers.
Acknowledgements
We are delighted to dedicate this paper in honor of Professor José Luis Gázquez. Financial assistance from CSIR, New Delhi and the Indo-EU (HYPOMAP) project is gratefully acknowledged. We are thankful to Dr. S Bandaru for helpful discussion. PKC would like to thank DST, New Delhi for the J. C. Bose National Fellowship.
References
1. Lewis, N.S.; Nocera, D.G. Proc. Natl. Acad. Sci. U.S.A. 2006, 103, 15729-15735. [ Links ]
2. Lubitz, W.; Tumas, W. Chem. Rev. 2007, 107, 3900-3903. [ Links ]
3. Cohen, R. L.; Wernick, J. H. Science 1981, 214, 1081-1087. [ Links ]
4. Graetz, J. Chem. Soc. Rev 2009, 38, 73-82. [ Links ]
5. Lochan, R. C.; Head-Gordon, M. Phys. Chem. Chem.Phys. 2006, 8, 1357-1370. [ Links ]
6. http://www1.eere.energy.gov/hydrogenandfuelcells/. [ Links ]
7. Deng, W.-Q.; Xu, X.; Goddard, W. A. Phys. Rev. Lett. 2004, 92, 166103-166106. [ Links ]
8. Kim, G.; Jhi, S.-H. J. Phys. Chem. C 2009, 113, 20499-20503. [ Links ]
9. Cheng, H.; Sha, X.; Chen, L.; Cooper, A. C.; Foo, M.-L.; Lau, G. C.; Bailey, W. H., III; Pez, G. P. J. Am. Chem. Soc. 2009, 131, 17732-17733. [ Links ]
10. Froudakis, G. E. Nano Lett. 2001, 1, 179-182. [ Links ]
11. Wu, X.; Gao, Y.; Zeng, X. C. J. Phys. Chem. C 2008, 112, 8458-8463. [ Links ]
12. Li, Y.; Zhou, G.; Li, J.; Gu, B.-L.; Duan, W. J. Phys. Chem.C 2008, 112, 19268-19271. [ Links ]
13. Baldé, C. P.; Hereijgers, B. P. C.; Bitter, J. H.; de Jong, K. P. Angew. Chem. Int. Ed. 2006, 45, 3501-3503. [ Links ]
14. Grochala, W.; Edwards, P. P. Chem. Rev. 2004, 104, 1283-1316. [ Links ]
15. Lee, H.; Lee, J.-W.; Kim, D. Y.; Park, J.; Seo, Y.-T.; Zeng, H.; Moudrakovski, I. L.; Ratcliffe, C. I.; Ripmeester, J. A. Nature 2005, 434,743-746. [ Links ]
16. Florusse, L. J.; Peters, C. J.; Schoonman, J.; Hester, K. C.; Koh, C. A.; Dec, S. F.; Marsh, K. N.; Sloan, E. D. Science 2004, 306, 469-471. [ Links ]
17. Chattaraj, P. K.; Bandaru, S.; Mondal, S. J. Phys. Chem. A 2011, 115, 187-193. [ Links ]
18. Hashimoto, S.; Sugahara,T.; Sato, H.; Ohgaki, K. J. Chem. Eng. Data 2007, 52(2), 517-520. [ Links ]
19. Züttel, A.; Borgschulte, A.; Orimo, S. Scripta Mater. 2007, 56, 823-828. [ Links ]
20. Weitkamp, J.; Firtz, M.; Ernst, S. Int J. Hydrogen Energy 1995, 20, 967-970. [ Links ]
21. Reilly, J. J. Hydrogen Storage and their Applications; CRC Press: Cleveland, OH, 1977, Vol. 2, p 13. [ Links ]
22. Orimo, S.; Nakamori, Y.; Eliseo, J. R.; Züttel, A.; Jensen, C. M. Chem. Rev. 2007, 107, 4111-4132. [ Links ]
23. Han, S. S.; Deng, W.-Q.; Goddard III, W. A. Angew. Chem., Int. Ed. 2007, 46, 6289-6292. [ Links ]
24. Furukama, H.; Miller, M. A.; Yaghi, O. M. J. Mater. Chem. 2007, 17, 3197-3204. [ Links ]
25. Collins D. J.; Zhou H.-C. J. Mater. Chem. 2007, 17, 3154-3160. [ Links ]
26. Cabria, I.; Lopez, M. J.; Alonso, J. A. Phys. Rev. B 2008, 78, 075415. [ Links ]
27. Kuc, A.; Zhechkov, L.; Patchkovskii, S.; Seifert, G.; Heine, T. Nano Lett. 2007, 7, 1-5. [ Links ]
28. Wu, H.; Zhou, W.; Yildirim, T. J. Am. Chem. Soc. 2007, 129, 5314-5315. [ Links ]
29. Rankin, R. B.; Liu, J.; Kulkarni, A. D.; Johnson, J. K. J. Phys. Chem. C 2009, 113, 16906-16914. [ Links ]
30. Zhou, M.; Wang, Q.; Zhang, L.; Liu, Y.-C.; Kang, Y. J. Phys. Chem. B 2009, 113, 11049-11053. [ Links ]
31. Sheppard, D. A.; Buckley, C. E. Int J. Hydrogen Energy 2008, 33, 1688-1692. [ Links ]
32. Klontzas E.; Mavrandonakis V.; Tylianakis E.; Froudakis G. E. Nano Lett. 2008, 8, 1572-1576. [ Links ]
33. Blomqvist A.; Moyses Araujo C.; Srepusharawoot P.; Ahuja R. Proc. Natl. Acad. Sci. 2007, 104, 20173-20176. [ Links ]
34. Srinivasu, K.; Ghosh, S. K. J. Phys. Chem. C 2010, 115, 1450-1456. [ Links ]
35. (a) Pan, S.; Giri, S.; Chattaraj, P. K. J. Comp. Chem. 2012, 33, 425-434. [ Links ] (b) Pan, S.; Merino, G.; Chattaraj, P. K. Phys. Chem. Chem. Phys. 2012, 14, 10345-10350. [ Links ] (c) Das, R.; Chattaraj, P. K. J. Phys. Chem. A 2012, 116, 3259-3266. [ Links ]
36. Chattaraj, P. K.; Roy, D. R.; Elango, M.; Subramanian, V. J. Phys. Chem. A 2005, 109, 9590-9597. [ Links ]
37. Chatt, J.; Manojlovic-Muir, L.; Muir, K. W. J. Chem. Soc D., Chem. Commun. 1971, 655-656. [ Links ]
38. Parkin, G. Acc Chem Res 1992, 25, 455-460. [ Links ]
39. Parkin, G. Chem. Rev. 1993, 93, 887-911. [ Links ]
40. Parkin, G.; Hoffmann, R. Angew. Chem., Int. Ed. 1994, 106, 1530. [ Links ]
41. Hoffmann, R.; Schleyer, P. v. R.; Schaefer, H. F., III. Angew. Chem. Int. Ed. 2008, 47, 7164-7167. [ Links ]
42. Giri, S.; Abhijith Kumar, R. P. S.; Chakraborty, A.; Roy, D. R.; Duley, S.; Parthasarathi, R.; Elango, M.; Vijayraj, R.; Subramanian, V.; Merino G.; Chattaraj, P. K. "Bonding, Aromaticity and Possible Bond-Stretch Isomerism in an "All-Metal" Cluster - [Be6Zn2]2- " in Aromaticity and Metal Clusters ed. P. K. Chattaraj, Taylor & Francis: Florida, (2009). [ Links ]
43. Parr, R. G.; Yang, W. Density Functional Theory of Atoms and Molecules, Oxford University Press: New York, 1989. [ Links ]
44. Chattaraj, P. K. (ed.) Chemical Reactivity Theory: A Density Functional View; Taylor & Francis/CRC Press: Florida, 2009. [ Links ]
45. Geerlings, P.; De Proft, F.; Langenaeker, W. Chem. Rev. 2003, 103, 1793-1873. [ Links ]
46. Chattaraj, P. K.; Giri, S. Ann. Rep. Prog. Chem. Sect. C: Phys. Chem. 2009, 105, 13. [ Links ] 240 J. Mex. Chem. Soc. 2012, 56(S)
47. Sen, K. D.; Jorgenson, C. K. Eds. Structure and Bonding, Vol. 66: Electronegativity; Springer: Berlin, 1987. [ Links ]
48. Chattaraj, P. K. J. Indian. Chem. Soc. 1992, 69, 173-184. [ Links ]
49. Sen, K. D.; Mingos, D. M. P. (eds.) Structure and Bonding, Vol. 80: Chemical Hardness; Springer: Berlin, 1993. [ Links ]
50. Parr, R. G.; Pearson, R. G. J. Am. Chem. Soc. 1983, 105, 7512-7516. [ Links ]
51. Pearson, R. G. Chemical Hardness: Applications from Molecules to Solids; Wiley- VCH: Weinheim, 1997. [ Links ]
52. Parr, R. G.; Szentpaly, L. v.; Liu, S. J. Am. Chem. Soc. 1999, 121, 1922-1924. [ Links ]
53. Chattaraj, P. K.; Sarkar, U.; Roy, D. R. Chem. Rev. 2006, 106, 2065-2091. [ Links ]
54. Chattaraj, P. K.; Roy, D. R. Chem. Rev. 2007, 107, PR46 - PR74. [ Links ]
55. Mulliken, R. S. J. Chem. Phys. 1955, 23, 1833-1840. [ Links ]
56. (a) Pearson, R. G. J. Chem. Ed. 1987, 64, 561-567. [ Links ] (b) Gázquez, J. L. J. Phys. Chem. A 1997, 101, 4657-1659 [ Links ]
57. Parr, R. G.; Chattaraj, P. K. J Am. Chem. Soc. 1991, 113, 1854-1855. [ Links ]
58. Ayers, P. W.; Parr, R. G. J. Am. Chem. Soc. 2000, 122, 2010-2018. [ Links ]
59. Chattaraj, P. K.; Sengupta, S. J. Phys. Chem. 1996, 100, 16126-16130. [ Links ]
60. Fuentealba, P.; Simon-Manso, Y.; Chattaraj, P. K. J. Phys. Chem. A 2000, 104, 3185-3187. [ Links ]
61. Chamorro, E.; Chattaraj, P. K.; Fuentealba, P. J. Phys. Chem. A 2003, 107, 7068-7072. [ Links ]
62. Parthasarathi, R.; Elango, M.; Subramanian, V.; Chattaraj, P. K. Theo. Chem. Acc. 2005, 113, 257-266. [ Links ]
63. Koopmans, T. A. Physica, 1933, 1, 104-113. [ Links ]
64. Janak, J. F. Phys. Rev. B, 1978, 18, 7165-7168. [ Links ]
65. Gaussian 03, Revision B. 03, Gaussian, Inc., Pittsburgh, PA. [ Links ]
66. Schleyer, P.v.R.; Maerker, C.; Dransfeld, A.; Jiao, H.; Hommes, N.J.R.V.E. J. Am.Chem. Soc. 1996, 118, 6317-6318. [ Links ]
67. Kumar, T. J. D.; Tarakeshwar, P.; Balakrishnan, N. Phys. Rev. B 2009, 79, 205415. [ Links ]
68. Chattaraj, P. K.; Duley, S. J. Chem. Eng. Data 2010, 55, 1882-1886. [ Links ]
69. Chakraborty, A.; Giri, S.; Duley, S.; Anoop, A.; Bultinck, P.; Chattaraj, P. K. Phys. Chem. Chem. Phys. 2011, 13, 14865-14868. [ Links ]
70. Zubarev, D. Y.; Boldyrev, A. I. J. Phys. Chem A 2008, 112, 7984-7985. [ Links ]
* The asterisk indicates the name of the author to whom inquires about the paper should be addressed.

