










Services on Demand
Journal
Article
 English (pdf)
English (pdf)
 Article in xml format
Article in xml format Article references
Article references
 Send this article by e-mail
Send this article by e-mailIndicators
-
 Cited by SciELO
Cited by SciELO -
 Access statistics
Access statistics
Related links
-
 Similars in
SciELO
Similars in
SciELO
Share

 Permalink
PermalinkJournal of the Mexican Chemical Society
Print version ISSN 1870-249X
J. Mex. Chem. Soc vol.56 n.2 Ciudad de México Apr./Jun. 2012
Article
Diligustilide: Enantiomeric Derivatives, Absolute Configuration and Cytotoxic Properties
Alejandra León and Guillermo Delgado*
Instituto de Química de la Universidad Nacional Autónoma de México, Circuito Exterior, Ciudad Universitaria, Coyoacán 04510, México, D.F. *delgado@unam.mx
Received January 24, 2012.
Accepted June 4, 2012.
Abstract
New enantiomeric amides (–)–6, (+)–7, (+)–6, and (–)–7 were formed by the reaction of the natural dimeric phthalide rac–diligustilide (rac–1) with (R)–(+)–α–methylbenzylamine and (S)–(–)–α–methylbenzylamine. The absolute configurations of compounds 6 and 7 were assigned by the analysis of electronic circular dichroism curves by means of the exciton chirality method. Compounds 1, 4, 5, (–)–6, (+)–6, (+)–7, and (–)–7 exhibited cytotoxic activity towards several human tumor cell lines.
Key words: Ligusticum porteri, diligustilide, enantiopure derivatives, electronic circular dichroism, exciton chirality method, cytotoxicity.
Resumen
Las amidas enantioméricas novedosas (–)–6, (+)–7, (+)–6 y (–)–7 fueron preparadas por medio de la reacción de la ftálida dimérica natural rac–diligustílida (rac–1) con (R)–(+)–a–metilbencilamina y (S)–(–)–α–metilbencilamina. Las configuraciones absolutas de los compuestos 6 y 7 fueron asignadas por medio del análisis de las curvas de dicroísmo circular electrónico mediante el método de la quiralidad del excitón. Los compuestos 1, 4, 5, (–)–6, (+)–6, (+)–7 y (–)–7 mostraron actividad citotóxica frente a varias líneas celulares de tumores humanos.
Palabras clave: Ligusticum porteri, diligustílida, derivados enantiopuros, dicroísmo circular electrónico, método de la quiralidad del excitón, citotoxicidad.
Introduction
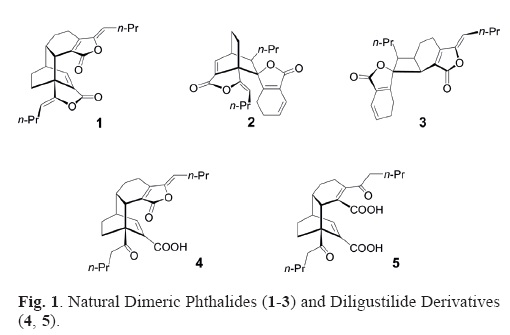
The rhizome of Ligusticum porteri C. & R. (known as "oshá") has been used in traditional medicine to treat colds, sore throats and stomachaches [1], and infusions are employed in ritual curing ceremonies by the Raramuri and the Zuni Indians [2]. The main components of L. porteri are Z–ligustilide, Z–butyli–denephthalide, ferulic acid, rac–diligustilide (1), rac–tokinolide B (2), and rac–riligustilide (3) among other constituents [3,4]. Rac–diligustilide (1) and rac–tokinolide B (2) have been obtained via [π4s + π2s] cycloaddition using Z–ligustilide as diene and dienophile [5, 6], and the acid catalyzed reaction of the monomer afforded linear dimeric products [7]. The chemical reactivity of rac–diligustilide has been the subject of several studies. Treatment under basic conditions of rac–diligustilide (1) afforded products with intramolecular carbon–carbon and oxygen–carbon connectivities [8]. The hydrolysis in basic media of 1 yielded a mixture of demethylwallichilide (4) and the diketo diacid of rac–diligustilide (5) [6]. Base catalyzed treatment of rac–diligustilide (1) under several reactions conditions afforded intramolecular condensation derivatives [9]. Recently, we reported the preparation of enantiomeric derivatives of rac–tokinolide B (2), determined their absolute configurations and evaluated their cytotoxic activity [10]. In this context, we were interested in obtaining enantiomeric derivatives of rac–diligustilide (1) that could be evaluated in some human cancer cell lines as cytotoxic agents.
Results and Discussion
As an initial study for the preparation of enantiomerically pure derivatives of rac–diligustilide (1), we examined different reaction conditions to prepare the diastereomeric salts from the carboxylic acids, rac–demethylwallichilide (4) and the rac–diketo diacid of diligustilide (5) with enantiomerically pure amines, and (S)–(–)–α–methylbenzylamine. However, these reactions were unsuccessful, since lactonization (to produce rac–1) is a competitive process under these reaction conditions. Therefore, we undertook direct attempts to transform the natural product rac–1 with optically active amines. Fortunately, treatment of rac–diligustilide (1) in toluene under reflux with enantiomerically pure (. )–(+)– and (S)–(–)–α–methylbenzyl–amine led to a mixture of diastereomeric amides [(–)–6 + (+)–7 and (+)–6 + (–)–7, respectively] (Scheme 1) in a convenient preparative yield. The mixture of products from each reaction was separated by chromatographic procedures to obtain pure compounds as colorless oils. The stereochemical relationships were confirmed by their spectroscopic properties, the specific rotations and by the ECD curves. The stereoisomers (–)–6, (+)–7, (+)–6, and (–)–7 had molecular formulae C32H39O4N, established by FAB–HR–MS, indicating the addition of the en–antiomerically pure amines (C8H11N) to rac–diligustilide (1) (C24H28O4). The IR spectra exhibited absorption bands for three carbonyl groups [1763, 1705, 1656 cm–1 for (–)–6 and (+)–6; 1768, 1704, 1658 cm–1 for (+)–7 and (–)–7]. The 32 carbon signals observed in the 13C NMR spectra for each structure were assigned by DEPT analysis to three carbonyl groups: a ketone [δC 208.5, C–3'], an α,β,γ,δ–unsaturated–γ–lactone [δC 169.0, C–1 for (–)–6 and (+)–6; 169.2, for (+)–7 and (–)–7], and the amide conjugated with an endocyclic double bond [δC 168.6, C–1' for (–)–6 and (+)–6; 167.7, C–1' for (+)–7 and (–)–7]; one quaternary carbon [δC 152.8, C–3a for (–)–6 and (+)–6; 153.0, C–3a for (+)–7 and (–)–7]; and a benzylic methyl group [δC 21.7, C–9" for (–)–6 and (+)–6; 21.3 C–9" for (+)–7 and (–)–7]. The change in the chemical shift for C–1', a new signal for C–3' for the keto group, and the absence of the double bond C3'/C8' were evidences for the structure of the products. The signals for the moieties of the (R)–(+)– and (S)–(–)–α–methylbenzylamine for each product were identified (see Experimental Section). The new signals in the 1H NMR spectra at δH 5.45, H–1" for (+)–6 and (+)–6; 5.68, H–1" for (+)–7 and (–)–7; and 5.05, H–2" for (–)–6 and (+)–6; 5.08 H–2", for (+)–7 and (–)–7 confirmed the addition of the (R)–(+)– and (S)–(–)–α–methylbenzylamine to the starting material, securing the structures (–)–6, (+)–6, (+)–7, and (–)–7. The mechanism of the transformation involves opening of the γ–lactone fused to the bicycle [2.2.2] via a nucleo–philic attack of the enantiomerically pure amine at its carbonyl to afford the corresponding amides.
The absolute configurations of the diastereomeric amides (–)–6, (+)–6, (+)–7, and (–)–7 were determined by the interpretation of the electronic circular dichroism (ECD) with the assistance of the exciton–chirality method [11–13] using the preferred conformations computed by energy minimization [14].
The experimental ECD spectra for compounds (+)–6, (–)–6, (+)–7, and (–)–7 were rather complicated due to the overlapof several exciton couplets of the different chromophores (the α,β,γ,δ–unsaturated–γ–lactone, the ketone group, the α,β–unsatu–rated amide and the benzene moiety). To facilitate the interpretation of the ECD spectra, and particularly, for the identification of the spatial arrangement of the chromophores, we considered the Newman projections through five a bonds taking C–7 at the front and C–2" at the back as the extremes (Figures 2A and 2B for compound (–)–6 of the preferred conformation [14]. C–7 and C–2" are chiral carbons vicinal to the chromophores.

In the low energy region of the CD of the levorotatory (and major) product of the reaction of rac–1 with (R)–(+)–α–methylbenzylamine (Figure 3, curve a) were observed two Cotton effects [n→π* 304 nm (Δε+2.19); π→π*267 (Δε–5.25)] which were weaker in intensity than those transitions attributed to the α, β, γ, δ,–unsaturated γ–lactone and benzene moieties. The first one was negative [237 nm (Δε–11.46)] and the second one was positive [206 nm (Δε+7.27)], defining a negative chirality, therefore, this curve was assigned to structure (–)–6 in agreement with the spatial disposition of the two chromo–phores for this structure (Figures 2C,2D), establishing the absolute stereochemistry 6. , 7. , 3'a. , 6'. , 2". for this compound (Figure 2).

On the other hand, (+)–7 (the minor product of the reaction of rac–1 with (R)–(+)–α–methylbenzylamine) exhibited pseudoe–nantiomeric ECD with respect to (–)–6 (Figure 3b). The Cotton effect signals of the ketone and amide group [n→π*, 302 nm (Δε –1.87) and π—π*, 271 nm (Δε+4.42)] were weaker with respect to the absorption at 219 nm (Δε +16.34) corresponding to the α, β, γ, δ–unsaturated γ–lactone. The second effect was positive [205 nm (Δε +11.4)], therefore, the spatial orientation of the transition dipoles of the chromophores (α,β,γ,δ–unsaturated γ–lactone and the benzene moiety) is clockwise. Considering the Newman projection through C–7/C–2" (Figure 4A,B), this clockwise contribution (Figure 4 C, D) defined the 6S, 7S, 3'aS, 6'S and 2". configuration for (+)–7.
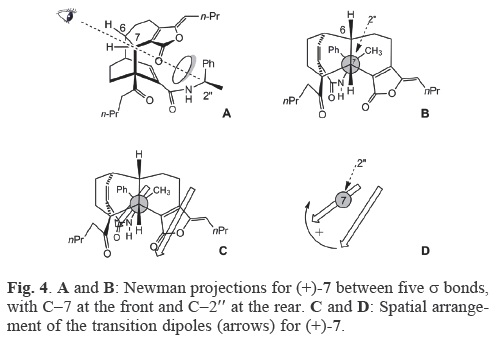
The Cotton effect of the dextrorotatory (and major) product obtained for the reaction of rac–1 with (S)–(–)–α–methylben–zylamine [271 nm (Δε +11.96) and 203 nm (Δε –8.23)] was enantiomeric with respect to that of (–)–6 and showed positive chirality, establishing 6S, 7S, 3'aS, 6'S and 2"S as the absolute configuration for (+)–6. Complementary to this, (–)–7 exhibited only one ECD extreme at 272 nm (Δε –11.35) as the first Cotton effect, while the second effect was buried in a strong negative background ellipticity. A negative contribution of the benzene moiety and the α,β,γ,δ–unsaturated γ–lactone chromo–phores established the absolute configuration 6R, 7R, 3'aR. , 6'R. and 2"S for (–)–7 (Scheme 1).
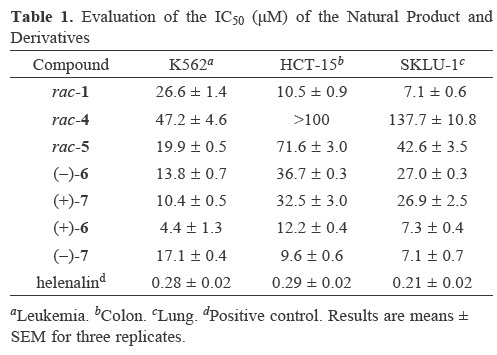
Considering the pharmacological importance of the dimeric phthalides and their potential as cytotoxic agents [10,15,16], we tested the isolated derivatives against three human cancer cell lines following standard protocols [10,17]. The IC50 values are shown in Table 1. The results indicated that the natural product rac–diligustilide (1) showed the best activity compared with its carboxylic derivatives rac–4 and rac–5. There are differences in the bioactivities of the enantiomers, with (+)–6 being approximately 3–fold more active than (–)–6 and (–)–7 being approximately 3–fold more active than (+)–7 with the exception of the K562 line. While the enantiomeric derivatives (–)–6, (+)–7, (+)–6 y (–)–7 were promising, they were also nonselective and displayed only marginally better inhibitory activity than the parent compound toward the cell lines tested.
These results confirm the unique features of the chemistry of dimeric phthalides [3–10]. In this case, only the 1,2–nu–cleophilic addition of the chiral amine to one lactone was observed, confirming the relative stability of the α,β,γ,δ–unsaturated γ–lactone of rac–1, and no intramolecular reactions were observed. The differences in bioactivity of the enantiomeric lactams indicate the chiral nature of the targets.
Experimental Section
General Experimental Procedures
Rac–diligustilide (1) was isolated from the acetone extract of the rhizomes of Ligusticum porteri by column chromatography [3], carried out on silica gel (230–400 mesh, Merck). Thin layer chromatography analyses were done on aluminum–backed silica gel 60 F254 plates (0.20 mm thickness) plates (Merck) and visualization of chromatograms were under UV lamp and then with solution of ammonium cerium sulfate. Infrared spectra were recorded with FTIR Bruker TENSOR 27 instrument. Ultraviolet spectra were determined on a Shimadzu UV160U Instrument. The optical rotation was measured in MeOH using a Perkin–Elmer 341 polarimeter. The 1H and 13C NMR experiments were performed at 25 °C using Varian UnityPlus 500 spectrometer (at 500/125 MHz), the spectra were recorded in CDCl3 (7.26 and 77.0 ppm, for 1H and 13C NMR, respectively) as reference. The chemical shifts (5) are expressed in ppm relative to TMS, and the coupling constants (J) in Hz. EIMS and HRMS (FAB+) spectra were recorded on a JEOL SX102A mass spectrometer. The (R)–(+)– and (S)–(–)–α–methylbenzylamine Chiraselect > 99.0% (Sum of enantiomers, GC) were purchased from Fluka Sigma–Aldrich.
Preparation of rac–demethylwallichilide (4) and rac–diketodiacid of diligustilide (5)
Demethylwallichilide (4) [6] and rac–2iketo2iaci2 of diligusti–lide (5) [9] were prepared following the procedures previously described.
Treatment of 4 with (R)–(+)–α–methylbenzylamine
Demethylwallichilide (4, 10 mg, 0.025 mmol) was dissolved in EtOAc (10 mL) and (R)–(+)–α–methylbenzylamine (6 (μL, 0.04 mmol) was then added. The reaction mixture was stirred at room temperature and then refluxed for 5 h. After usual work up, a yellow oil was obtained which was purified by TLC preparative (n–heptane/EtOAc, 2:3) to afford rac–diligustilide (1) as the main product (5 mg, 52.4%) and starting material (2 mg, 20%) was recovered.
Treatment of 5 with (S)–(–)–α–methylbenzylamine
5 (25 mg, 0.06 mmol) was dissolved in MeOH (10 mL) and (S)–(–)–α–methylbenzylamine (11 (μL, 0.09 mmol) was added, the mixture was stirring at room temperature for 1 h, then was refluxed for 5 h. After usual work up, only the starting material was recovered.
Derivatization of rac–Diligustilide (1) with (R)–(+)–α–methylbenzylamine
To a solution of rac–diligustilide (4, 99.2 mg, 0.26 mmol) in dry toluene (10 mL) was added (R)–(+)–α–methylbenzylamine (57.12 mg, 0.47 mmol), the reaction mixture was stirred and refluxed under a nitrogen atmosphere for 20 h. The reaction mixture was cooled, neutralized with HCl (10%), extracted with EtOAc (3 x 10 mL), and the organic layers were joined and washed with brine, dried with Na2SO4, and the solvent evaporated under reduce pressure. The residue (yellow oil, 115.3 mg) was purified by preparative TLC (n–hexane/EtOAc 7:3, developing three times) recovering 7.9% of starting material and affording 92.54 mg (70.8%) of two products:
(–)–6 (43.18 mg, 33%) as a colorless oil; Rf 0.50 (n–hex–ane/EtOAc, 3:2, twice); [α]25D –96.1 (c 1.3 x 10–3, MeOH); UV (MeOH) λmax (log ε): 276 (3.6), 208 (4.3); CD (c 1.68 x 10–5, MeOH): 304 nm (Δε +2.19), 267 nm (Δε –5.25), 237 nm (Δε –11.46), 206 nm (Δε +7.27); IR (CHCl3) vmax: 3435, 3012, 2961, 2937, 2874, 1763, 1705, 1656, 1496, 1451, 1378, 1233, 1167, 1133, 997 cm–1; 1H NMR (CDC13, 500 MHz; assignments by COSY, NOESY and HMQC) δ 7.30 (2H, 222, J = 8.5, 8.5, 1.0 Hz, H–5", H–7"), 7.27 (2H, 22, J = 8.5, 1.5 Hz, H–4", H–8"), 7.22 (1H, 2222, J = 8.5, 8.5, 1.5, 1.5, H–6"), 6.50 (1H, 2, J = 6.5 Hz, H–7'), 5.45 (1H, 2, J = 8.0 Hz, H–1"), 5.09 (1H, t, J = 7.5 Hz, H–8), 5.05 (1H, 2222, J = 7.5 Hz, H–2"), 3.19 (1H, 2, J = 8.5 Hz, H–7), 2.88 (1H, 222, J = 18.5, 7.5, 7.5 Hz, H–8'a), 2.60–2.53 (2H, m, H–6', H–8'b), 2.46–2.44 (1H, m, H–6), 2.31 (2H, 2222, J = 13.0, 7.5, 7.5, 7.5 Hz, H–9a, H–9b), 2.23 (1H, t, J = 4.5 Hz, H–4a), 2.19–2.16 (1H, m, H–4b), 2.14 (1H, 222, J = 12.5, 6.0, 3.0, 3.0 Hz, H–4'a), 1.88 (1H, 2222, J = 15.5, 6.5, 3.0, 3.0 Hz, H–5a), 1.81 (1H, 2222, J = 14.0, 8.5, 4.0, 4.0 Hz, H–5'a), 1.70–1.63 (3H, m, H–4'b, H–9'a, H–9'b), 1.63–1.58 (1H, m, H–5'b), 1.45 (2H, q, J = 7.5 Hz, H–10a, H–10b), 1.46 (1H, 2, J = 6.5 Hz, H–9"), 1.43–1.40 (1H, m, H–5b), 1.36 (2H, 2222, J = 15.0, 7.5, 7.5, 7.5 Hz, H–10'a, H–10'b), 0.93 (3H, t, J = 7.0 Hz, H–11), 0.92 (3H, t, J = 7.5 Hz, H–11'); 13C NMR (CDCl3, 125 MHz; assignments by DEPT, HSQC and HMBC) δ 208.5 (C–3'), 169.0 (C–1), 168.6 (C–1'), 152.8 (C–3a), 148.5 (C–3), 143.8 (C–3"), 141.8 (C–7'a), 135.6 (C–7'), 128.5 (C–5", C–7"), 128.1 (C–7a), 127.0 (C–6"), 126.1 (C–4", C–8"), 111.2 (C–8), 57.3 (C–3'a), 48.6 (C–2"), 39.4 (C–7), 39.2 (C–8'), 37.9 (C–6'), 37.6 (C–6), 29.4 (C–4'), 28.5 (C–5'), 28.0 (C–5), 27.9 (C–9), 25.4 (C–9'), 22.4 (C–10), 22.4 (C–10'), 21.7 (C–9"),19.4 (C–4), 14.1 (C–11'), 13.8 (C–11); EIMS m/z 501 [M+] (23), 461 (11), 444 (4), 381 (7), 311 (25), 226 (8), 191 (100), 120 (98), 105 (77), 79 (10), 57 (9), 43 (6), 29 (5); HRMS (FAB+) m/z 502.2955 (calcd for C32H39O4N+H+ 502.2957).
(+)–7 (49.36 mg, 37.7%) as a colourless oil; Rf 0.45 («–hex–ane/EtOAc, 3:2, twice); [α]25D +117.0 (c 1.35 x 10–3, MeOH); UV (MeOH) λmax(log ε): 277 (3.5), 207.8 (4.3); CD (c 2.4 x 10–5, MeOH): 302 nm (Δε –1.87), 271 nm (Δε +4.42), 219 nm (Δε +16.34), 205 nm (Δε +11.40); IR (CHCl3) vmax: 3436, 3012, 2961, 2874, 1768, 1704, 1658, 1496, 1450, 1377, 1232, 1167, 1133, 997, 920 cm–1; 1H NMR (CDC13, 500 MHz; assignments by COSY, NOESY and HMQC) δ 7.31 (2H, ddd, J = 7.5, 7.5, 1.5 Hz, H–5", H–7"), 7.35 (2H, 22, J = 8.5, 1.5 Hz, H–4", H–8"), 7.23 (1H, 2222, J = 8.5, 7.0, 1.5, 1.5, H–6"), 6.58 (1H, 2, J = 6.5 Hz, H–7'), 5.68 (1H, 2, J = 8.0 Hz, H–1"), 4.99 (1H, t, J = 8.0 Hz, H–8), 5.08 (1H, 2222, J = 7.0 Hz, H–2"), 3.29 (1H, b2, J = 9.0 Hz, H–7), 2.95 (1H, 222, J = 15.0, 6.0, 6.0 Hz, H–8'a), 2.59–2.56 (1H, m, H–8'b), 2.56–2.53 (1H, m, H–6'), 2.48–2.44 (1H, m, H–6), 2.28 (2H, q, J = 7.5, Hz, H–9a, H–9b), 2.22 (1H, q, J = 7.5 Hz, H–4a), 2.14–2.07 (2H, m, H–4b, H–4'a), 1.89 (1H, 2222, J = 15.5, 6.5, 3.5, 3.5 Hz, H–5a), 1.79 (1H, 2222, J = 14.0, 8.5, 3.5, 3.5 Hz, H–5'a), 1.69–1.54 (4H, m, H–4'b, H–5'b, H–9'a, H–9'b), 1.34 (2H, q, J = 7.5 Hz, H–10a, H–10b), 1.47 (1H, 2, J = 6.5 Hz, H–9"), 1.43–1.40 (1H, m, H–5b), 1.34 (2H, q, J = 7.5 Hz, H–10'a, H–10'b), 0.92 (3H, t, J = 7.5 Hz, H–11), 0.89 (3H, t, J = 7.5 Hz, H–11'); 13C NMR (CDCl3, 125 MHz; assignments by DEPT, HSQC and HMBC) δ 208.5 (C–3'), 169.2 (C–1), 167.7 (C–1'), 153.0 (C–3a), 148.4 (C–3), 143.2 (C–3"), 141.5 (C–7'), 136.5 (C–7'), 128.5 (C–5", C–7"), 127.6 (C–7a), 127.0 (C–6"), 126.4 (C–4", C–8"), 111.0 (C–8), 57.2 (C–3'a), 48.3 (C–2"), 39.4 (C–7), 39.0 (C–8'), 37.9 (C–6'), 37.8 (C–6), 29.8 (C–4'), 28.4 (C–5'), 28.2 (C–5), 27.9 (C–9), 25.6 (C–9'), 22.4 (C–10), 22.5 (C–10'), 21.3 (C–9"), 19.2 (C–4), 14.0 (C–11), 13.8 (C–11'); EIMS m/z 501 [M+] (17), 461 (6), 444 (4), 381 (5), 341 (7), 311 (16), 227 (10), 191 (73), 105 (100), 79 (12), 60 (55), 43 (94), 29 (19), 18 (24); HRMS (FAB+) m/z 502.2961 (calcd for C32H39O4N+H+ 502.2957).
Derivatization of rac–Diligustilide (1) with (S)–(–)–α–methylbenzylamine
To a solution of rac–diligustilide (1, 100 mg, 0.26 mmol) in dry toluene (10 mL) was added (S)–(–)–α–methylbenzylamine (63.7 mg, 0.52 mmol), the reaction mixture was stirred and refluxed under a nitrogen atmosphere for 20 h. The reaction mixture was cooled, then neutralized with a solution of HCl (10%) and extracted with EtOAc (3 x 10 mL), the organic layers were joined, washed with brine, dried with Na2SO4, and the solvent was evaporated under reduced pressure. The residue (yellow oil, 120 mg) was applied in a preparative TLC (n–hexane/EtOAc 4:1, developing four times) recovering 6.3% of 4 and affording 92.36 mg (75.6%) of two products:
(+)–6 (48.13 mg, 39.5%) as a colourles oil; Rf 0.56 (n–hexane/EtOAc, 2:3, twice); [α]25D +46.3 (c 9.5 x 10–4, MeOH); UV (MeOH) λ,max (log ε): 276.5 (3.8), 207.5 (4.3); CD (c 2.2 x 10–5, MeOH) 302 nm (Δε –4.80), 271 nm (Δε +11.96), 243 nm (Δε +8.01), 203 nm (Δε –8.23); HRMS (FAB+) m/z 502.2954 (calcd for C32H39O4N+H+ 502.2957).
(–)–7 (44.23 mg, 36.3%) as a colourless oils; Rf 0.54 (n–hexane/EtOAc, 2:3, twice); [α]25D –125.8 (c 8.5 x 10–4, MeOH); UV (MeOH) λmax (log e): 278 (3.9), 207 (4.3); CD (c 1.9 x10–5, MeOH) 303 nm (Δε +4.47), 272 nm (Δε –11.35), 218 nm (Δε –11.82), 203 nm (Δε –12.50); HRMS (FAB+) m/z 502.2956 (calcd for C32H39O4N+H+ 502.2957).
Cytotoxicity assay
Colon (HCT–15), leukemia (K–562), and lung (SKLU–1) human tumor cell lines were supplied by National Cancer Institute (NCI), USA. The cytotoxicity of the test compounds was 2etermine2 using the protein–binding 2ye sulforhodamine B (SRB) [17]. The IC50 values (mean ± standard error for three replicates) are shown in Table 1.
Acknowledgments
Financial supports from CONACyT (scholarship 181999 and project 102158) and Programa de Posgrado en Ciencias Químicas de la UNAM are gratefully acknowledged. We thank the technical assistance of M. I. Chávez, B. Quiroz–García, M. T.
References
1. Bye, R.; Linares, E. J. Ethnobiol. 1986, 6, 289–306. [ Links ]
2. Linares, E.; Bye, R. J. Ethnopharmacol. 1987, 19, 153–183. [ Links ]
3. León, A.; Toscano, R. A.; Tortoriello, J.; Delgado, G. Nat. Prod. Res. 2011, 25, 1234–1242. [ Links ]
4. León, A.; Chávez, M. I.; Delgado, G. Magn. Reson. Chem. 2011, 49, 469–476. [ Links ]
5. Ogawa, Y.; Mori, Y.; Maruno, M.; Wakamatsu, T. Heterocycles 1997, 45, 1869–1872. [ Links ]
6. Rios, M. Y.; Delgado, G.; Toscano, R. A. Tetrahedron 1998, 54, 3355–3366. [ Links ]
7. Rios , M. Y.; Delgado, G. J. Mex. Chem. Soc. (former Rev. Soc. Quím. Méx.) 1999, 43, 127–132. [ Links ]
8. Rios, M. Y.; Delgado, G.; Espinosa–Pérez, G. Tetrahedron Lett. 1998, 39, 6605–6608. [ Links ]
9. Quiroz–García, B.; Hernández–Ortega, S.; Sterner, O.; Delgado, G. Tetrahedron 2004, 60, 3681–3688. [ Links ]
10. León, A. Cogordán, J. A.; Sterner, O.; Delgado, G. J. Nat. Prod. 2012, 75, 859–864. [ Links ]
11. Harada, N.; Nakanishi, K. Acc. Chem. Res. 1972, 5, 257–263. [ Links ]
12. Harada, N.; Nakanishi, K. Circular Dichroic Spectroscopy–Exciton Coupling in Organic Stereochemistry; University Science Books, Mill Valley, CA, and Oxford University Press, Oxford, 1983. [ Links ]
13. Boiadjiev, S. E.; Lightner, D. A. Monatshefte für Chemie 2005, 136, 482–509. [ Links ]
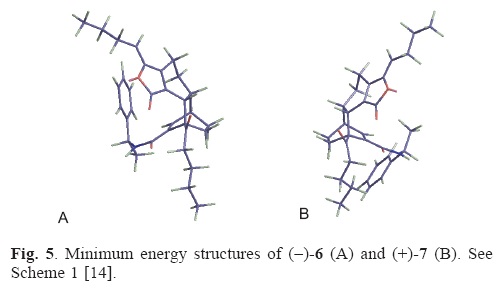
14. Energy minimization was performed with MM2 force field using ChemBio3D Ultra 12.0. 1986–2009 Cambridge Soft Corporation. The preferred conformations of (–)–6 (Figure 5A) and (+)–7 (Figure 5B) showed similar features that included an extended conformation of the n–propyl chains, a pseudo half boat conformation of the cyclohexene ring fused with the bicyle [2.2.2], a sec– trans arrangement for the α,β–unsaturate2 lactam and a syn–relationship between the amide nitrogen and the benzylic methine.
15. Lin, G.; Chan, S. S. K.; Chung, H. S.; Li, S. L. In Studies in Natural Products Chemistry, Ed., Atta–ur–Rahman, Elsevier: Amsterdam, 2006; Vol. 32. Bioactive Natural Products Part L, pp. 611–669. [ Links ]
16. Beck, J. J.; Chou, S. J. J. Nat. Prod. 2007, 70, 891–900. [ Links ]
17. Monks, A.; Scudiero, D.; Skehan, P.; Shoemaker, R.; Paull, K.; Vistica, D.; Hose, C.; Langley, J.; Cronise, P.; Vaigro–Wolff, A.; Gray–Goodrich, M.; Campbell, H.; Mayo, J.; Boyd, M. J. Natl. Cancer Inst. 1991, 83, 757–766. [ Links ]

