










Servicios Personalizados
Revista
Articulo
 Inglés (pdf)
Inglés (pdf)
 Artículo en XML
Artículo en XML Referencias del artículo
Referencias del artículo
 Enviar artículo por email
Enviar artículo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO -
 Accesos
Accesos
Links relacionados
-
 Similares en
SciELO
Similares en
SciELO
Compartir

 Permalink
PermalinkJournal of the Mexican Chemical Society
versión impresa ISSN 1870-249X
J. Mex. Chem. Soc vol.56 no.2 Ciudad de México abr./jun. 2012
Article
Implementing Stepwise Solvent Elution in Multisyringe Liquid Chromatography (MSC) to Separate Norepinephrine, Epinephrine and Dopamine
María del Pilar Cañizares-Macías,1'* Hilda María González-San Miguel,2 and Víctor Cerdà2
1 Facultad de Química, Departamento de Química Analítica, Universidad Nacional Autónoma de México, México D.F., 04510, México. *pilarm@unam.mx
2 Department of Chemistry, University of the Balearic Islands, Carretera de Valldemossa Km. 7.5, E-07122 Palma de Mallorca, Spain.
Received September 11, 2011.
Accepted April 4, 2012.
Abstract
A multisyringe liquid chromatographic (MSC) method by using stepwise solvent elution and reverse-phase liquid chromatography C18 monolithic column to separate Epinephrine (E), Norepinephrine (NE) and Dopamine (DA) was developed. The first step employs the mixture sodium1-hexanosulfonate monohydrate (HS) 7.5mM: methanol (98:2) to separate NE and E and the second step uses HS (5.0 mM): methanol (92:8) to elute DA. The three analytes were separated on 6 min spending only 10 mL of mobile phases. The detection limits were between 1.3 μg mL-1 (DA) and 2.04 μg mL-1 (NE).
Keywords: Multisyringe liquid chromatography; C18 monolithic column; catecholamines; stepwise elution.
Resumen
Se desarrolló un método por cromatografía líquida multijeringa (MSC) para separar Epinefrina (E), Norepinefrina (NE) y Dopamina (DA) por elutión en varios pasos acoplando una columna monolítica C18 en fase reversa. Para la primera etapa se utilizó una mezcla de 1-hexanosulfonato de sodio heptahidratado (HS) 7.5 mM: metanol (98:2) para separar E y NE y para eluir DA se utilizó una mezcla HS 5.0 mM:metanol (92:8). Los tres anlitos se separaron en 6 min con un gasto total de las fases móviles de 10 mL. Los límites de detectión estuvieron entre 1.3 μg mL-1 (DA) y 2.04 μg mL-1 (NE).
Palabras clave: Cromatografía Líquida Multijeringa; Columna monolítica; catecolaminas.
Introduction
Continuous flow techniques have improved in the last decades. The first published papers for non-segmented systems appeared in the 57' and its evolution has been exponential, so that they have been chosen as the main tools to automatize the analytical methods. The versatility of these systems has allowed to couple dialysis modules [1, 2], immobilized enzymes [3, 4] or ionic exchange columns [5], so it has been possible to analyze more than one compound at the same time.
Sequential Injection Analysis (SIA) developed by Ruzicka y Marshall in 1990 [6], is one of the more versatile flow technique because allows the selection of the reagents and sample by a multiposition valve using bidirectional propulsion system [7, 8]. SIA has been later coupled with monolithic columns, allowing the development of the Sequential Injection Chromatography (SIC), which permits the separations by low and medium pressure liquid chromatography in a fast way [9, 10].
Multisyringe Flow Injection Analysis (MSFIA) has also showed great potential and versatility in the analysis of complex samples since the monolithic columns have also been successfully coupled to this system obtaining the Multisyringe Liquid Chromatography (MSC) [12], mixing the versatility of the continuous flow systems with the separation capacity of the chromatography. MSC allows saving organic solvents and money, avoiding the use the expensive HPLC instruments [11-13]. From the beginning, MSC has been used with different kinds of samples showing its robustness, selectivity and sensitivity. It has been used to separate b-lactam antibiotics [14] or to determine the chemiluminescence of thiazide compounds in urine samples coupling on-line the isolation and preconcentra-tion of the analytes by means of a solid phase extraction [15].
MSC is coming out as an alternative to the HPLC in the determination of several pharmaceutical compounds with the same precision, selectivity and efficiency.
Norepinephrine (NE), Epinephrine (E) and Dopamine (DA) are biogenic amines based on a benzene ring with hydroxyl groups at 3- and 4-positions, and with an amino group in a side chain. Due to the fact that the benzene ring with the two vicinal groups is called a catechol ring, the name catecholamines were given to these substances (Fig. 1).
Catecholamines act via dopaminergic and adrenergic receptors, and are involved in a variety of regulatory systems. They play an important role in the central nervous system as neurotransmitters or hormones, and they also affect the regulation blood and the regulation of the response to stress [16-19]. Due to presence of many catecholaminergic pathways and a wide range of functions in the central nervous system a development of realibles techniques for their quantification is essential. The HPLC method combined with electrochemical (EC) detection and a C18 reversed-phase HPLC column is still considered the method of choice. For the resolution of the catecholamines different mobile phases have been probed using principally a mixture water-methanol at different ratios. Buffer salts and ion paring agent (acetate 0.049 M, citric acid 0.019 M, EDTA 1.075 x 10–4 M) [20], or 0.05 M sodium acetate, 0.15 M acetic acid, 0.01 % (w/v) of sodium dodecyl sulfate (SDS), 0.01 % (w/v) sodium chloride and 0.01 % (w/v) EDTA are used as mobile phases to improve the separation [21]. Acetonitrile has been also used as modifier with 0.025M sodium phosphate, 0.025M citric acid, 0.001M heptane and sulphonic acid pH 2.85 [22]. HPLC analysis allows a good resolution of catecholamines with short time of analysis but the equipment is very expensive. Liquid chromatography coupled to mass spectrometry or tandem mass spectrometry detectors also constitutes an excellent analytical methodology for low concentrations of catecholamines and metanephrines. In the last few years some innovative methods have been reported [23-26], with high analytical performance for catecholamines determination based on fluorescence and chemiluminescence detection. Although sensitive, these methods are expensive and usually they do not allow remote data acquisition.
Other techniques and methods as capillary electrophoresis, that has showed a high sensitivity using different detection systems (UV, laser induced fluorescence, luminescence, etc) [27-30] or a fluorescent sensor applying derivative synchronous fluorescence spectrometry [31] have been also used to quantify catecholamines in different samples.
In this paper a new method by MSC using a stepwise solvent elution to separate NE, E and DA in mixtures of pharmaceutical samples has been developed. Stepwise solvent elution allows a chromatographic separation more efficient based on the isocratic elution using different mobile phases, so the mobile phases are introduced to the chromatography system in different steps. In flow systems stepwise elution has been implemented exploring the commutation of rotary injection/selection valve or the multi-syringe concept using more than two solvent composition [32]. Using chromatographic separation in combination with multisyringe flow analysis further enhanced the simplicity and the low cost of flow-based methods with the required selectivity or even specificity [33].
Results and discussion
Separation conditions
Selection of the mobile phase
In accordance with the references [34-36] mixtures of aceto-nitrile/methanol/buffer (pH between 3 and 7.5) with different ratios were evaluated as mobile phases but the three compounds overlapped and the separation was not possible.
It is known that when a reverse phase column is used, ion pairing agents improve the separation of polar analytes [20,21]. The addition of HS as ion pairing was evaluated adjusting in all solutions pH at 3 with phosphates in accordance with references for the HPLC determination of catecholamines using an ion pairing [37,38]. With 2.5 mM and 5 mM HS concentrations contained in mobile phase the peaks of the three compounds were much tailed with width peaks up to 180 s. So methanol, as an organic modifier, was used. When a HS 5 mM:methanol (95:5) mixture was used as mobile phase the peak purity of the three compounds were better but also the DA peak overlapped with the E peak, and NE and E was not separated with a good resolution (Rs<0.5). So, the concentration of HS was increased up to 7.5 mM to improve the resolution and the percentage of methanol was decreased down at 1 % to avoid overlapping of DA. The NE and E separation improved but the peaks were more width. By increasing of the concentration of methanol at 2 % the NE and E peaks were very well defined and were partly separated (Rs =1). DA was solved but its peak was much width eluting much later than E and NE so it was necessary to fill twice the syringe.
In order to get the DA peak purity and to elute it faster, an increase of the ratio of methanol and a decrease of HS in the mobile phase were carried out. The concentration of methanol was increased up to 8 % in order to get a more shaped peak; methanol percentages higher caused uncontrolled bubbles in the system. So, with the HS (5mM):methanol (92:8) mobile phase (MP2) a standard of 100 ug vaL1 of DA eluted in 4 min but this mobile phase was not suitable to separate E and NE. These were successfully separated using the MPj (HS (7.5 mM):methanol (98:2)).
So, as the optimum mobile phase to separate and elute the three catecholamines was different (MP1 and MP2), a stepwise gradient elution was chosen for their resolution.
Stepwise solvent elution
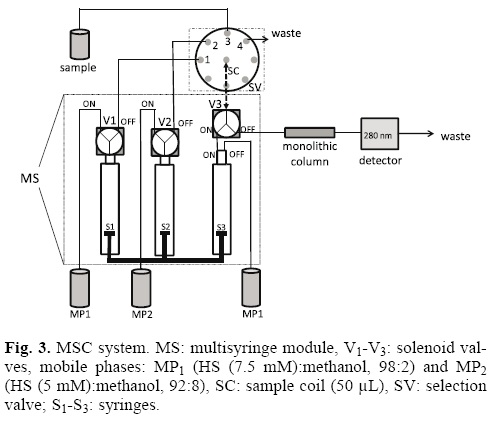
In stepwise solvent gradient mode different mobile phases are used to elute the analytes into the chromatographic system in an independent way. In this method the advantage is that the best mobile phase can be used for each analyte improving the separation. So, the separation of the three catecholamines was done using a stepwise solvent gradient mode. Two mobile phases were used: MP1 (HS (7.5. mM):methanol (98:2) to separate NE and E, and MP2 (HS (5 mM) methanol (92:8) to elute DA. Hydrodinamics parameter such as injection flow-rate, for both mobile phases, and injection volume were evaluated because the dispersion effect is more accused by these parameters when low pressure flow systems are used [12,33].
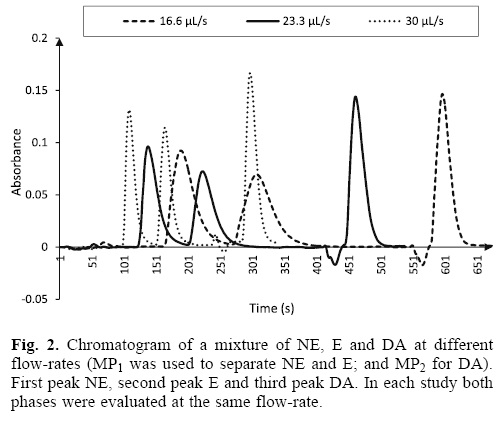
Flow-rate and injection volume: the MSC system is low pressure and controlled as by the commutation valves of the syringes in the multisyringe module as by the solenoid valves. The monolithic columns allow a 33.33 μL s_1 flow-rate and the MSC can get this flow-rate, but is better to work with low flow-rates to prolong the life of syringes and valves. Two solenoid valves of 600 kPa (V1 and V3) and one of 200 kPa (V2) were used. High flow-rates caused less dispersion and a shorter analysis time. According to Cerdà, the symmetry of peaks increases with decreasing the flow-rate as a consequence of diffusion effect [12]. NE and E are no easy separated because its structures are very similar (Fig. 1) therefore the evaluation of the hydrodynamic factors was carried out with the aim of improving their separation, although DA was also studied. The factorial design showed that flow-rate and injection volume are key in the model with a P-value lower than 0.05, thus indicating that these variables are significantly different from zero at 95.0 % confidence level, so these conditions have a significant effect on the peak area. Table 1 shows the values found of sum of square and mean square for NE and E. The test showed that when both values increased the peak area was higher. Besides when the flow-rate increased dispersion was lower. Fig. 2 shows the chromatograms obtained with different flow-rates for a mixture of NE, E and DA. Therefore, the separation of NE and E was carried out with a flow-rate of 30μL s_1 and DA was elutedto a flow-rate of 23.3μL s_1 because V2 solenoid valve —which connects the MP2 to the system—, had only 200 kPa. Flow-rate higher was not tested to avoid a system overpressure.
With regard to the injection volume the dispersion phenomena and resolution of NE and E were key to select the optimum value. Although with 200 μL the signal was higher for the three analytes, the peaks were wider. With volumes lower than 20 μL the peaks height was very small limiting the sensibility of the method. With a volume of 100 μL the peak area was high for the three analytes but the resolution (Rs) for NE and E was 1.10. When the volume was decreased down to 50 μL the area also decreased but the Rs increased up to 1.45. So 50 μL of injection volume was selected as optimum.
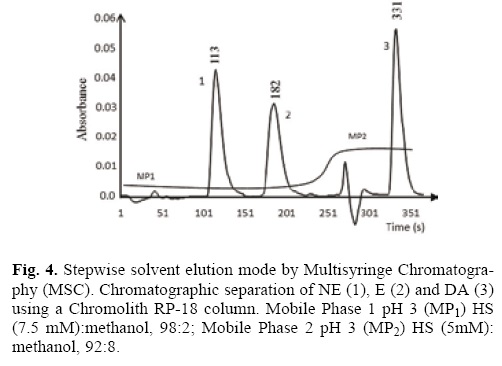
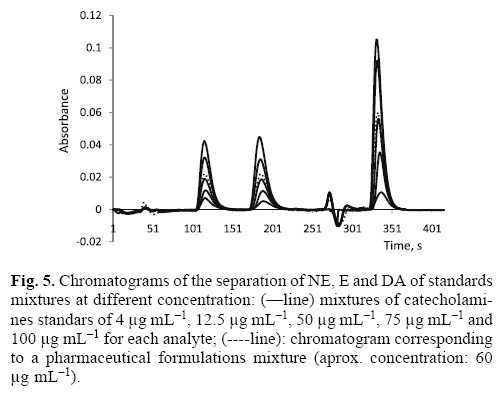
Once the chromatographic and hydrodynamics parameters were evaluated and according to the sequence of Table 2 and Fig. 3, the three analytes were separated in 6.1 min. The retention time for NE was 1.88 min (113 s), for E was 3.03 min (182 s) and for DA was of 5.51 min (331 s) as it is shown in the chromatogram of Fig. 4.
Analytical features of the proposed method
Calibration curves were built and the precision of the method was calculated using mixtures of the three catecholamines. Table 3 shows the calibrations curves for each analyte and the detection limit, reproducibility and repeatability of the method. The method shows a good sensibility for the three analytes with regression coefficient higher than 0.999. The method showed a very good precision both reproducibility and repeatability but the detection limits were high because the UV detection is not very sensitive. For the separation of catecholamines by HPLC several detectors have been used such as electrochemical, fluorescence or mass spectrometry, which detect very low concentration (ng mL1 or less). When UV detection is used the reported concentrations are at μg mL1 or μmol L_1 levels [39,40]. With the MSC proposed the found concentrations were between 4 μg mL1 (12 μmol L_1) and 80 μg mL1 (240 (μmol L_1) for E, NE and for DA, expressed as (imol L_1, between 21 and 421.
Analysis of pharmaceutical formulations mixtures
The method was applied to pharmaceutical formulations. They were analyzed by making a mixture of the three samples. Fig. 5 also shows the separation pharmaceutical mixtures (dot line), which is equal to the separation of different standard mixtures of the three compounds (straight line). Table 4 shows the obtained mean concentration in the pharmaceutical formulations which had values very closed to the reported by the manufacturer (±1 %). The error calculated for each concentration is expressed as μg mL–1 in accordance with its standard deviation relative. Recoveries between 99 % and 102 % were found when a concentration of 40 μg mL–1 of each analyte was added to the samples demonstrating the precision and accuracy of the method.
The cost of the MSC equipment is cheaper than HPLC besides it is a totally automated system, fast, precise and with a little waste, So, the proposed method shows the versatility of the multisyringe systems and the possibility to be used in separation and quantification of catecholamines as an alternative to HPLC.
Conclusions
In this paper norepinephrine, epinephrine and dopamine were separated at first time using a multisyringe chromatography system coupling a monolithic column and stepwise solvent elution mode. The system allows, using solenoid valves, reaching a very good efficiency even at low pressure, so excellent results were obtained in the separation of the three analytes at different concentrations with a very good precision (between 1.6 % and 3.5 %).
The method has allowed that the separation could be carried out using a stepwise solvent elution using only 10 mL of mobile phase: 6.60 mL for MP1 and 3.35 mL for MP2. In the other hand, this separation mode is a good choice for the elution and the separation of catecholamines because it has allowed using the best mobile phase in according to the characteristics of each compound.
The novel methodology capitalizes on the combination between flexibility and simplification for the separation of these catecholamines with analysis time and spending reagents similar to HPLC.
MSC exploit the ability of monolithic columns of relatively large pore sizes to effect separations without the need for high-pressure pumps. By contrast, HPLC can use not only monolithic columns, but also columns packed with very small particles through which fluids can be propelled only with the aid of high-pressure pumps.
One immediate comparison of the two techniques can be made in terms of the way that the mobile phase or eluent is released. While all two techniques allow switching to a different eluent at any time, so far, only HPLC uses a gradient for this purpose. MSC affords in-line switching to a different eluent (or mixture of eluents) by using at least two of the four syringes available. This novel operational mode allows successive isocratic release of several mobile phases. MSC therefore allows the mobile phase to be managed in ways not available with HPLC. Rapid switching between eluents in HPLC is unusual, since particle-packed columns require a gradual change to reequilibrate and provide a steady baseline. The monolithic columns used in MSC are more tolerant of eluent switching and equilibrate more rapidly, so the use of equipment allowing the eluent to be changed or its composition altered in a fairly speedy manner, which, as stated above, is relatively easy in MSC. HPLC is a more efficient technique; also, it allows gradients to be used in the separation of analytes in complex samples, and the high-pressure it employs usually involves a quaternary mixer capable of mixing eluents at low pressure and then propelling the resulting mixture at high pressure.
One other difference between the two techniques is in how samples are injected. Sample injection in MSC can be done in other ways (e.g., with a selection valve or several three-way valves). By contrast, HPLC requires using a dedicated device for injection; also, although the injection mechanism is accurate and reproducible, it considerably increases equipment costs relative to MSC.
Experimental
Reagents and mobile phases
(-)-Epinephrine(+)bitartrate salt, (E); L-(-)-Norepinephrine bitartrate salt monohydrate (NE) and Dopamine hydrochloride (DA) were purchased from Sigma-Aldrich (analytical grade, St. Louis, MO, USA). Two stock solutions of 1000 μg mL1 and 100 ug mL1 of E, NE and DA in 0.01 M HC1 (Scharlau, Barcelona, Spain) were prepared and stored under 4 °C in amber glass flasks. Working mixtures were prepared by proper dilution of the stock solutions with a 0.1 M pH 3 phosphate buffer. All the solutions were prepared with ultra-pure Millipore quality water (Millipore Corp. New York, USA).
Elution of analytes was achieved using two mobile phases prepared with the following composition: the Mobile Phase 1 (MP1 (pH 3) consisted of 0.1 M H3P04/KH2P04 buffer, 7.5 mM HS mixed with 2 % (v/v) methanol and the Mobile Phase 2 (MP2) (pH 3) consisted of 0.1 M H3P04/KH2P04 buffer, 5 mM HS mixed with 8 % (v/v) methanol. These were filtered through a Nylon membrane of 0.45 μm (Millipore) and degassed by ultrasound for about 15 min prior to use. Methanol (HPLC grade) was purchased by Scharlau (Barcelona, Spain) and sodium-1-hexanesulfonate monohydrate (HS) by Fluka (St. Louis, MO, USA).
MSC instrumentation
The MSC system was built with a multisyringe automatic burette (Crison; Alella, Barcelona, Spain) used to drive the liquid to the flow network, three solenoid valves: two MTV-3-N1(4UKG) and one MTV-2-l/4(UGH) (Takasago, Japan) and one automatic selection valve (Crison) used as the interphase to fill the sample and to select the two mobile phases.
The chromatographic separation was carried out using a Chromolith Flash RP-18e, (25mm><4.6mm i.d.) column protected with a Chromolith RP-18e (10mmx4.6mmi.d.) guard column (Merck). To build the manifold, poly(tetrafluoroethy lene) (PTFE) and peek tubing of 0.5 mm and of 0.8 mm of internal diameter were used. A Hewlett Packard 8453 diode array spectrophotometer equipped with an 18 ul inner volume flow-cell (Hellma) was used as detector. The catecholamines were measured at 280 nm. The Autoanalysis 5.0 software (Sci-ware, Palma de Mallorca. Spain) was used to control all instruments (burette, valves and detector) and for data logging and treatment [33].
MSC proposed method
A mixture of the three analytes was used to evaluate the separation of NE, E and DA using two mobile phases. In Fig. 3 the used MSC manifold is shown. The burette module uses only one stepper motor, which simultaneously moves the three syringes. Depending on the position of the solenoid valves, the liquids are picked up (PK) or dispensed (DP) to the manifold or to the reservoirs. Syringe Sj (10 mL) and syringe S2 (10 mL) were used to manage MPj and MP2, respectively. Syringe S3 (5 mL) was used to load and transfer the sample using a commutation valve and a solenoid one. To load the sample, 50 uL were aspired through the selection valve (SV) (position 3) towards the sample coil (SC), being V3 and S3 in the ON position and the system in the PK position. The system was in DP position and S3 and V3 in OFF position to transfer the sample to the monolithic column RP-18 (MC), the position of the SV, V1 and V2 valves depended on the selected mobile phase. The system was fixed in DP position and MP was introduced to it changing SV to position 1, Vj to position OFF and V2 to position ON to separate NE and E. Once these two analytes were eluted, SV was changed to position 2 and MP2 was passing through with V2 in position OFF and V in position ON for, at last, to allow DA to be eluted out of the monolithic column. During the elution of the three analytes V3 was kept in position OFF. Table 2 shows a brief summary of the analytical procedure used.
Study of the mobile phases properties
E, NE and DA are amines with different structures: DA and NE are primary amines and E is a secondary amine. The main difference between NE and DA is that the last one does not have an OH group in the aliphatic chain (Fig. 1).
Firstable, in accordance with the references [34-36], different ratios of acetonitrile/methanol/buffer with pH between 3 and 7.5 (pH 7.5 is the highest value when we are working with this kind of column) were also tested.
On the other, hand mobile phases containing HS at different concentrations (2.5 mM-7.5 mM), adjusted at pH 3 with phosphate buffer 0.1 M, and methanol (1 %-8%) were studied to select the composition of the best mobile phases.
All the experimental for the selection of the mobile phases were done applied an isocratic separation mode using two standard concentrations (100 μg mL-1 and 40 μg mL1) of each catecholamine and injected 100 μL. The MSC system was similar to Fig. 3 without using the S2 syringe: to aspirate the sample, the system was switched to position PK, the selection valve to position 3, the V3 valve and the S3 syringe to position ON and V to position ON. To inject and separate the analytes was switched to position dispensed (DP); V valve to position OFF, V3 and S3 to position OFF and SV to position 1. In these tests the S1 and S3 syringes reservoirs were filled with the mobile phase that was being tested.
Evaluation of the hydrodynamic parameters of the MSC system
The hydrodynamic conditions of the system were selected by using a factorial design 22. The evaluated factors were: flow-rate (minimum value 8.33 uL s_1 for both mobile phases and maximum value 30 μL s_1, for MP1 and 25 μL s_1 for MP2 because V2 solenoid valve —which connects the MP2 to the system—, had only 200 kPa) and injection volume at 20 μL and at 200 μL. The injection volume was set with the sample coil (SC) length. An ANOVA test was used to determine the influential factors. The optimum conditions of separation were selected from the analysis time, the wide and the high of the peaks and the analytes resolution (Rs).
Analytical performance evaluation: calibration study
The standard solutions of mixtures of the three catecholamines were prepared in phosphate buffer 0.1 M pH 3 and used for calibration studies, performing six replicates of the various standard concentrations (from 1 μg mL1 to 200 μg mL-1) of each catecholamine. In order to evaluate the precision of the method, within-laboratory reproducibility and repeatability were evaluated in a single experimental set-up with duplicates. Two measurements of a mixture of each standard (60 μg mL1 of each analyte) per day were carried out on seven days. ANOVA test was used to carry out the evaluation.
Pharmaceutical formulations analysis
The used samples to validate the separation of the three cat-echolamines by MSC were pharmaceutical formulations:
Adrenaline Level: Syringe with one milliliter, each mil-liliter contains 1 mg adrenaline (epinephrine) base in aqueous solution, excipient: sodium metabisulphite, chlorobutanol, hydrochloric acid, NaCl.
Norages; Norepinephrine bitartrate salt monohydrate, injection solution (8mg/4 mL), contains 1002 mg of norepinephrine base per liter of solution.
Dopamine fides, dopamine hydrochloride, 200 mg/10 mL, injection solution; excipient: sodhplium metabisulphite and water.
An aliquot of each formulation was mixed and the mixture was diluted a volume suitable using a 0.1 M pH 3 phosphate buffer with the aim to separate of three analytes in one just injection.
All the samples were analyzed six times and a mean was calculated. A recovery study adding 40 μg mL1 to the diluted samples (20 μg mL1) were also carried out.
Acknowledgements
The corresponding author thanks the support of the Directión General de Asuntos del Personal Académico (DGAPA) of the Universidad Nacional Autónoma de México. This research was carried out with the financial assistance provided by the Spain's Ministry of Science and Innovation (MICINN) through project CTQ2007-64331.
References
1. Medina Alonso, G.; Carrasco Fuentes, M.; Cañizares Macías, M. P. Talanta 2005, 68, 292-297. [ Links ]
2. Fontenele, R. S.; Hidalgo P.; Gutz, I. G. R.; Pedrotti J. J. Talanta 2007, 72, 1017-1022. [ Links ]
3. Jin, J.; Muroga, M.; Takahashi, F.; Nakamura, T. Bioelectrochem. 2010,79, 147-151. [ Links ]
4. Ketterer, L.; Keusgen, M. Anal. Chim. Acta 2010, 673, 54-59. [ Links ]
5. Di Nezio, M. S.; Palomeque, M. E.; Fernandez Band, B. S. Anal. Lett. 2006, 39, 1211-1228. [ Links ]
6. Ruzicka, J.; Marshall, G. Anal. Chim. Acta 1990, 237, 329-343. [ Links ]
7. Ruzicka, J.; Giiebeli, T. Anal. Chem. 1991, 63, 1680-1685. [ Links ]
8. Lenehan, C. E.; Barnett, N. W.; Lewis, S. W. Analyst 2002, 127, 997-1020. [ Links ]
9. Satinsky, D.; Huclova, J.; Solich, P.; Karlicek, R. J. Chromatogr. A 2003, 1015, 239-244. [ Links ]
10. Satínsky, D.; Solich, P.; Chocholouš, P.; Karlícek, R. Anal. Chim. Acta 2003, 499, 205-214. [ Links ]
11. Cerdà, V.; Estela, J. M.; Forteza, R.; Cladera, A.; Becerra, E.; Altimira, P.; Sitjar, P. Talanta 1999, 50, 695-705. [ Links ]
12. González-San Miguel, H. M.; Alpízar-Lorenzo, J. M.; Cerdà-Martín, V. Talanta 2007, 72, 296-300. [ Links ]
13. Fernández, M.; González-San Miguel, H. M.; Estela, J. M.; Cerdà, V. Trends in Anal. Chem. 2009, 28, 336-346. [ Links ]
14. González-San Miguel, H. M.; Alpízar-Lorenzo, J. M.; Cerdà, V. Anal. Bioanal. Chem. 2007, 387, 663-671. [ Links ]
15. Maya, F.; Estela, J. M.; Cerdà, V. Talanta 2010, 80, 1333-1340. [ Links ]
16. Leite, O. D.; Lupetti, K. O.; Fatibello-Filho, O.; Vieira, I. C.; Barbosa, A. M. Talanta 2003, 59, 889-896. [ Links ]
17. Tsunoda, M. Anal. Bioanal. Chem. 2006, 386, 506-514. [ Links ]
18. Southwick, S. M.; Paige, S.; Morgan, C. A.; Bremner, J. D.; Krystal, J. H.; Charney, D. S. Semin. Clin. Neuropsychiatry 1999, 4, 242-248. [ Links ]
19. Durstewitz, D.; Kelc, M.; Gunturkun, O. J. Neurosci. 1999, 19, 2807-2822. [ Links ]
20. Bergquist, J.; Sciubisz, A.; Kaczor, A.; Silberring, J. J. Neuroscience Meth. 2002, 113, 1-13. [ Links ]
21. Smedes, F.; Kraak, J. C.; Poppe, H. J. Chromatogr. 1982, 231, 25-39. [ Links ]
22. Ferreira, F. D. P.; Silva, L. I. B.; Freitas, A. C.; Rocha-Santos, T. A. P.; Duarte, A. C. J. Chromatogr. A 2009, 1216, 7049-7054. [ Links ]
23. Mercolini, L., Gerra, G., Consorti, M., Somaini, L., Raggi, M. A. Talanta 2009, 78, 150-155. [ Links ]
24. Nalewajko, E., Wiszowata, A., Kojlo, A. J. Pharm. Biomed. Anal. 2007, 43, 1673-1681 [ Links ]
25. de Jong, W. H. A.; de Vries, E. G. E.; Wolffenbuttel, B. H. R.; Kema, I. P. J. Chromatogr. B 2010, 878, 1506-1512. [ Links ]
26. Chan, E. C. Y.; P. C. Rapid Commun. Mass. Spectrom. 2000, 14, 1959-1964. [ Links ]
27. Robert, F.; Bert, L.; Denoroy, L.; Renaud, B. Anal. Chem. 1995, 67, 1838-1844. [ Links ]
28. Siren, H; Karjalainen, U. J. Chromatogr. A 1999, 853, 527-533. [ Links ]
29. William, E. G.; O'Connor, T. Electrophoresis 2010, 31, 55-64. [ Links ]
30. Zhang, N.; Zhang, H. S.; Wang, H. Electrophoresis 2009, 30, 2258-2265. [ Links ]
31. Cañizares, M. P.; Luque de Castro, M. D. Anal. Chim. Acta 1995, 317, 335-341. [ Links ]
32. Rigobello-Masini, M.; Penteado, J. C. P.; Liria, C. W.; Miranda, M. T. M.; Masini, J. C. Anal. Chim. Acta 2008, 628, 123-132. [ Links ]
33. Becerra, E.; Cladera, A.; Cerdà, V. Lab. Rob. Autom. 1999, 11, 131-140. [ Links ]
34. Valet, P.; Saulnier-Blache, J. S. Ann. Endocrinol. 1999, 60, 167-174. [ Links ]
35. Patel, B. A.; Arundell, M.; Parker, K. H.; Yeoman, M. S.; O'Hare, D. J. Chromatogr. B 2005, 818, 269-276. [ Links ]
36. Ueyama, J.; Kitaichi, K.; Iwase, M.; Takagi, K.; Takagi, K.; Hasegawa, T. J. Chromatogr. B 2003, 798, 35-41. [ Links ]
37. Grossi, G. Bargossi, A., Lippi, A., Battistoni, R. Chromatogr. 1987, 24, 842-846. [ Links ]
38. Nalewajko, E., Wiszowata, A., Kojło, A. J. Pharmac. Biomed. Anal. 2007, 43, 1673-1681. [ Links ]
39. Strobel, G.; Werle, E.; Helfinger, H; Griebel, D:; Weicker H., Eur. J. Biochem. 1988, 176, 397-402. [ Links ]
40. Calderón Guzmán, D.; Hernández García, E.; Osnaya Brizuela, N.; Trujillo Jiménez F.; Barragán Mejía, G.; Juárez Olguín, H.; Santamaría del Ángel, D.; Nuñez, A. E.; Carmona Aparicio, L. Arch Pharm Res. 2010, 33, 1671-1677. [ Links ]

