










Servicios Personalizados
Revista
Articulo
 Inglés (pdf)
Inglés (pdf)
 Artículo en XML
Artículo en XML Referencias del artículo
Referencias del artículo
 Enviar artículo por email
Enviar artículo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO -
 Accesos
Accesos
Links relacionados
-
 Similares en
SciELO
Similares en
SciELO
Compartir

 Permalink
PermalinkJournal of the Mexican Chemical Society
versión impresa ISSN 1870-249X
J. Mex. Chem. Soc vol.56 no.2 Ciudad de México abr./jun. 2012
Article
Microwave–assisted High Diastereoselective Synthesis of α –Aminophosphonates under Solvent and Catalyst Free–conditions
Gaurao D. Tibhe,1 Miguel Ángel Reyes–González,1 Carlos Cativiela,2 and Mario Ordóñez1*
1 Centro de Investigaciones Químicas, Universidad Autónoma del Estado de Morelos, 62209 Cuernavaca, Morelos (México).
2 Departamento de Química Orgánica, ISQCH, Universidad de Zaragoza–CSIC, 50009 Zaragoza (Spain).
Received October 26, 2011.
Accepted April 2, 2012.
Abstract
A simple, efficient and general method has been developed for the high diastereoselective synthesis of α–aminophospho–nates through "one–pot" three–component reaction of alkyl and aryl aldehydes with (S)–α–methylbenzylamine or (S)–3,3–dimethyl–2–butylamine and dimethyl phosphite (Kabachnik–Fields reaction), which proceeds in short time using microwave irradiation under solvent and catalyst free–conditions. This method could be useful in the large–scale synthesis of α–aminophosphonates in short reaction time.
Key words: Three–component Reaction, Diastereoselective Synthesis, α–aminophosphonates, Aicrowave, Green–Chemistry.
Resumen
Se desarrolló un método eficiente, general y simple para la síntesis altamente diastereoselectiva de α–aminofosfonatos a través de una reacción "one–pot" de tres componentes de aldehídos alifáticos y aromáticos con (S)–α–metilbencilamine o (S)–3,3–dimetil–2–butilamina y fosfito de dimetilo (reacción de Kabachnik–Fields), la cual procede en tiempos cortos utilizando irradiación de microondas en ausencia de disolvente y catalizador. Este método podría ser de utilidad para la síntesis de α–aminofosfonatos a gran escala y en tiempos cortos de reacción.
Palabras clave: Reacción tres–componentes, síntesis diastereoselectiva, α–aminofosfonatos, microondas, química–verde.
Introduction
In the last two decades, the α–aminophosphonic acids 1 and their phosphonate derivatives have received considerable attention in synthetic organic and medicinal chemistry, since they are analogues of the natural a–amino acids 2, versatile intermediates and act as transition state mimics 3 during peptide bond hydrolysis.
The utility of the α–aminophosphonates as peptidomimetics, pharmacogenetic agents, antitumoral enzymatic inhibitors, haptens of catalytic antibodies, inhibitors of UDP–galactopyra–nose mutase and plant glutamine synthetase, antitumoral, antibiotics and pharmacologic agents are well documented [1,2]. For that reason, the synthesis of α–aminophosphonates has received considerable attention and significant progress has been made to develop more efficient methods for the synthesis of these compounds [3]. In this context, the "one–pot" three–component reaction (Kabachnik–Fields reaction) is one of the most useful methods for the synthesis of α–aminophosphonates due to its versatility and high yields. Recently, the "one–pot" three–component synthesis of α–aminophosphonates starting from aldehydes, amines and dialkyl or trialkyl phosphites has been reported using various catalysts such as A(ClO4)n [4], BF3.OEt2 [5], In(X)n [6], Bi(X)n [7], A(OTf)n [8], ACl3 [9], ZrOCl2 and H–beta zeolite [10], complex of tetra–tert–butyl–phthalocyanine aluminum chloride (t–PcAlCl) [11], yttria–zirconia [12], TiO2 [13], CdI2 [14], NbCl5 [15], (KAl(SO4)2–12H2O) [16], SiO2–AlCl3 [17], CuO [18], task–specific ionic liquid (TSIL) [19], I2 [20], [PyH]X [21], [bnmim][HSO4] [22], nano Fe3O4 [23], Al(H2PO4)3 [24], Yb(PFO)3 [25], PPh3 [26], tosyl chloride [27], TMSCl [28], and AcOH [29]. However, in spite of their potential utility, these procedures typically suffer from one or more disadvantages such as the use of expensive or less available or stoichiometric amount of catalyst, where specialized handling techniques and tedious work–up are necessary, as well as non–recyclability of the catalyst, long reaction time, vigorous reaction conditions, requirement of excess of reagent, use of solvent, unsatisfactory yields, and lack of generality. Consequently, there is still needs to develop a more efficient, simple, milder and high yield protocol. In this context, the "one–pot" three–component reaction under solvent and catalyst free–conditions [30–32] is an excellent alternative for the synthesis of α–aminophosphonates. Additionally, a combination of catalyst and ultrasonic [33] or microwave [34] irradiation leads to very strong acceleration of these reactions.
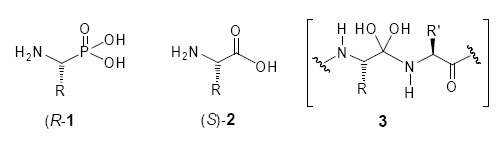
In view that the biological activity related to a–aminophos–phonic acids and derivatives depends on the absolute configuration of the stereogenic center a at the phosphorus atom [35], the development of a green approach for their stereoselective synthesis is desirable. In this context, and in connection with our green chemistry program, we recently reported an efficient and "one–pot" three–component procedure for the diastereose–lective synthesis of α–aminophosphonates. The reactions were carried out by heating the reagents (aldehyde, chiral amines and dimethyl phosphite) at 80 oC over a period of 5–8 h, depending on aldehyde structure [30]. In order to optimize the reaction conditions for the diastereoselective synthesis of both aliphatic and aromatic α–aminophosphonates in a short time, herein we report an eco–friendly, simple and efficient method based on a "one–pot" three–component reaction of aliphatic and aromatic aldehydes with the chiral amines 4–9 and dimethyl phosphite, using microwave–irradiation, and under solvent and catalyst free–conditions [36, 37].
Results and Discussion
We first investigated the "one–pot" three–component reaction of benzaldehyde with (S)–a–methylbenzylamine 4 [(S)–MBA] and dimethyl phosphite at 80 oC and 60 watts under solvent and catalyst free–conditions [38]. Under these conditions, we found that microwave irradiation causes a strong acceleration of this process (reaction time was shorten, going from 5–8 h to just a 12 min) to give the (R,S)– and (S,S)–α–aminophosphonates 10a in 81% yield as a 75:25 diastereoisomeric ratio (dr), which was determined according to their 31P NMR signals at 27.54 and 27.21 ppm, respectively. The stereochemistry of the α–aminophosphonates was established on the basis of our previous results [30], and by comparison with the results reported in the literature [4a, b].
After the optimization of the experimental conditions with the chiral amine (S)–α–MBA, 4, we extended this "one–pot" three–component reaction with other chiral amines such as (S)–4–me–thoxy–a–methylbenzylamine, 5, (S)–1–(1'–naphthyl)ethylamine, 6, (S)–1,2,3,4–tetrahydro–1–naphthylamine, 7, (R)–phenylgly–cinol, 8, and (S)–3,3–dimethyl–2–butylamine, 9. Thus, the reaction of benzaldehyde with the chiral amines 5–7 and dimethyl phosphite predominantly affords the (R,S)–a–aminophospho–nates 11a–13a (Table 1, entries 2–4). When (R)–phenylglycinol 8 was used as the chiral amine, (R,R)–α–aminophosphonate 14a was obtained as the major diastereoisomer (Table 1, entry 5) [4a, 29]. The three–component reaction of benzaldehyde with the chiral amines 5–8 and dimethyl phosphite proceeds with good chemical yields but with only moderate diastereoselectivi–ty. On the other hand, using the (S)–3,3–dimethyl–2–butylamine, 9 the reaction principally gave the (R,S)–α–aminophosphonate 15a in 86% yield and 89:11 dr (Table 1, entry 6). The stereochemistry of the obtained α–aminophosphonates 11a–15a was established by correlation of the spectroscopic data with those obtained for α–aminophosphonate 7a.
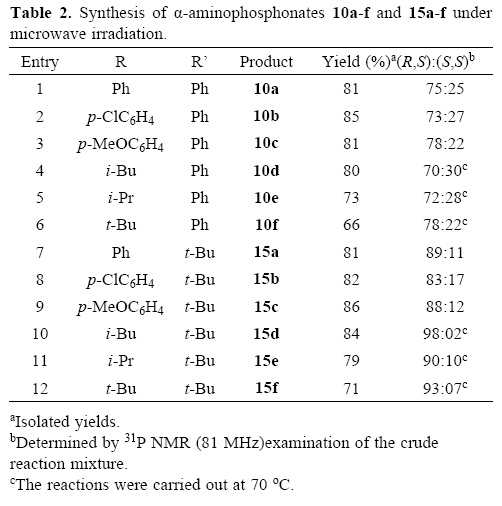
With these optimized conditions in hand, the "one–pot" three–component reaction of aliphatic and aromatic aldehydes with chiral amines 4 and 9 and dimethyl phosphite was carried out, obtaining the corresponding (R,S)– and (S,S)–a–ami–nophosphonates 10a–f (Table 2, entries 1–6) and 15a–f (Table 2, entries 6–12) with good yield and diastereoselectivity. A slight decrease in reaction rate was observed when aliphatic aldehydes were used.
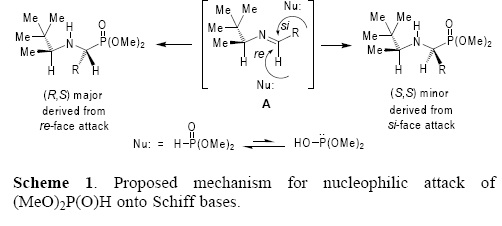
Based on our previous studies [30] and those reported in the literature [39], the origin of diastereoselectivity in the "one–pot" three–component reaction of aldehydes, (S)–3,3–dimethyl–2–butylamine 9 and (AeO)2P(O)H under microwave irradiation can be explained as illustrated in Scheme 1. The initial condensation reaction of aldehyde and 9 gave the corresponding Schiff base A, which adopts a conformation where the proton of the chiral fragment is eclipsed with the imine double bond, as should be expected from the 1,3–allylic strain model [40]. The conformation with the t–Bu–C– or Ae–C moieties eclipsed with N=C–H fragment are appreciably higher in energy as has been determined by ab initio MO and DFT studies. Consequently, the nucleophilic attack of dimethyl phosphite onto the Schiff base A takes place at the re face (less hindered face) to affords the α–aminophosphonates (R,S) as the major diastereoisomers.
In summary, we found a high diastereoselective "one–pot" three–component reaction of aldehydes, chiral amines and dimethyl phosphite under solvent and catalyst free–conditions using microwave irradiation. We also established that Schiff bases intermediates derived from (S)–3,3–dimethyl–2–butylamine 9 shows higher C=N π–facial selectivities than those found in the Schiff bases derived from commonly used (S)–a–methylbenzyl–amine 4 or the chiral amines 5–8. This procedure could be used in the synthesis of large amounts of α–aminophosphonates in short reaction times.
Experimental Section
General Information: All commercial reagents were used as received without further purification. Microwave reactions were performed in a CEM Discover System (with a power of 60 W). Flash chromatography was performed using 230–400 mesh Silica Flash 60® silica gel. Thin layer chromatography was performed with pre–coated TLC sheets of silica gel (60 F254, Aerck). NMR spectra were recorded with a Varian System instrument (400 MHz for 1H, and 100 MHz for 13C) and a Aercury instrument (81 AHz for 31P) and calibrated with CDCl3 as solvent and TMS as internal standard signal. Chemical shifts (δ) are reported in parts per million. Coupling constants (J) are given in Hz. High resolution FAB+ and CI+ mass spectra (HRMS) were obtained in a JEOL HRMStation JHRMS–700. Microanalyses were determined in an Elemental VARIO EL III machine.
General procedure for the synthesis of α–aminophosphonates under microwave irradiation: A mixture of aldehyde (1.0 equiv.) and chiral amine (1.0 equiv.) in an open flask without any solvent and catalyst was irradiated with AW (60 W) at 80 oC for 2 min. The flask was cooled at room temperature and dimethyl phosphite (1.05 equiv.) was added. The reaction mixture was again irradiated at 80 oC for 10 min. The crude products were analyzed by 31P NMR spectroscopy at 81 AHz, and then purified by column chromatography on silica gel, obtaining the corresponding (R,S)– and (S,S)–α–aminophosphonates. All spectroscopy data for α–aminophosphonates 10a–f and 15a–f have been reported by us [30].
Dimethyl (R,S)– and (S,S)–{(Phenyl)[(1–phenylethyl)am ino]methyl}phosphonate (10a): A mixture of benzaldehyde (250 mg, 2.35 mmol), (S)–α–MBA (280 mg, 2.35 mmol), and dimethyl phosphite (250 mg, 2.46 mmol) was irradiated at 80 oC for 12 min. Product 10a was obtained (601 mg, 81%) as a white solid; mp 60 0C.
Dimethyl (R,S)– and (S,S)–{(4–Chlorophenyl)[(1–phenylethyl)amino]methyl}phosphonate (10b): A mixture of 4–chlorobenzaldehyde (250 mg, 1.78 mmol), (S)–α–MBA (210 mg, 1.78 mmol), and dimethyl phosphite (200 mg, 1.87 mmol) was irradiated at 80 oC for 12 min. Product 10b was obtained (521 mg, 85%) as an oil.
Dimethyl (R,S)– and (S,S)–{(4–Methoxyphenyl)[(1–phe nylethyl)amino]methyl}phosphonate (10c): A mixture of 4–methoxybenzaldehyde (250 mg, 1.83 mmol), (S)–α–MBA (210 mg, 1.83 mmol), and dimethyl phosphite (200 mg, 1.92 mmol) was irradiated at 80 oC for 12 min. Product 10c was obtained (490 mg, 81%) as a yellow oil.
Dimethyl (R,S)– and (S,S)–{3–Methyl–1–[(1–phenylethyl )amino]butyl}phosphonate (10d): A mixture of isovaleralde–hyde (250 mg, 2.90 mmol), (S)–α–MBA (350 mg, 2.90 mmol), and dimethyl phosphite (330 mg, 3.04 mmol) was irradiated at 70 oC for 12 min. Product 10d was obtained (697 mg, 80%) as a yellow oil.
Dimethyl (R,S)– and (S,S)–{2–Methyl–1–[(1–phenylethyl)
amino]propyl}phosphonate (10e): A mixture of isobutyralde–hyde (250 mg, 3.46 mmol), (S)–α–MBA (410 mg, 3.46 mmol), and dimethyl phosphite (400 mg, 3.63 mmol) was irradiated by AW at 70 oC for 12 min. Product 10e was obtained (704 mg, 73%) as a yellow oil.
Dimethyl (R,S)– and (S,S)–{2,2–Dimethyl–1–[(1–phenyl ethyl)amino]propyl}phosphonate (10f): A mixture of tert–butylacetaldehyde (250 mg, 2.90 mmol), (S)–α–MBA (330 mg, 2.90 mmol), and dimethyl phosphite (330 mg, 3.04 mmol) was irradiated at 70 oC for 12 min. Product 10f was obtained (538 mg, 66%) as an oil.
Dimethyl (R,S)– and (S,S)–{(Phenyl)[(1,2,2–trimethy lpropyl)amino]methyl}phosphonate (15a): A mixture of Benzaldehyde (250 mg, 2.35 mmol), (S)–3,3–dimethyl–2–butyl–amine(237 mg, 2.35 mmol), and dimethyl phosphite (250 mg, 2.46 mmol) was irradiated at 80 oC for 12 min. Product 15a was obtained (568 mg, 81%) as a solid; mp 66 oC.
Dimethyl (R,S)– and (S,S)–{(4–Chlorophenyl)[(1,2,2–trimethylpropyl)amino]methyl}–phosphonate (15b): A mixture of 4–chlorobenzaldehyde (250 mg, 1.78 mmol), (S)–3,3–dimeth–yl–2–butylamine (179 mg, 1.78 mmol), and dimethyl phosphite (200 mg, 1.87 mmol) was irradiated at 80 oC for 12 min. Product 15b was obtained (640 mg, 82%) as an oil.
Dimethyl (R,S)– and (S,S)–{(4–Methoxyphenyl)[(1,2,2–trimethylpropyl)amino]methyl}–phosphonate (15c): A mixture of 4–methoxybenzaldehyde (250 mg, 1.83 mmol), (S)–3,3–dimethyl–2–butylamine (180 mg, 1.83 mmol), and dimethyl phosphite (200 mg, 1.92 mmol) was irradiated at 80 oC for 12 min. Product 15c was obtained (504 mg, 86%) as an oil.
Dimethyl (R,S)– and (S,S)–{7–Methyl–1–[(1,2,2–trimethy lpropyl)amino]butyl}phosphonate (15d): A mixture of iso–valeraldehyde (250 mg, 2.90 mmol), (S)–3,3–dimethyl–2–butyl–amine (290 mg, 2.90 mmol), and dimethyl phosphite (330 mg, 3.04 mmol) was irradiated at 70 oC for 12 min. Product 15d was obtained (671 mg, 84%) as an oil.
Dimethyl (R,S)– and (S,S)–{2–Methyl–1–[(1,2,2–trimethy lpropyl)amino]propyl}phosphonate (15e): A mixture of iso–butyraldehyde (250 mg, 3.46 mmol), (S)–3,3–dimethyl–2–butyl–amine (350 mg, 3.46 mmol), and dimethyl phosphite (400 mg, 3.53 mmol) was irradiated at 70 oC for 12 min. Product 15e was obtained (661 mg, 79%) as an oil.
Dimethyl (R,S)– and (S,S)–{2,2–Dimethyl–1–[(1,2,2–trimethylpropyl)amino]propyl}–phosphonate (15f): A mixture of tert–butylacetaldehyde (250 mg, 2.90 mmol), (S)–3,3–dimethyl–2–butylamine (490 mg, 2.90 mmol), and dimethyl phosphite (330 mg, 3.04 mmol) was irradiated at 70 oC for 12 min. Product 15f was obtained (961 mg, 71%) as an oil.
Acknowledgments
The authors thank CONACYT of México, for financial support via projects 62271 and J000.400/2009), Ministerio de Ciencia e Innovación (project CTQ2010–17436) and Consejo Superior de Investigaciones Científicas (project 2008AX0044. We thank to S. Lagunas–Rivera and V. Labastida–Galván for the determination of RMN and mass spectra. GDT and MARG also thank the CONACYT for a Graduate Scholarships.
References
1. (a) Orsini, F.; Sello, G.; Sisti, A. Curr. Med. Chem. 2010, 17, 264–289; [ Links ] (b) Naydenova, E. D.; Todorov, P. T.; Troev, K. D. Amino Acids 2010, 38, 23–30; [ Links ] (c) Lejczak, B.; Kafarski, P. Top. Heterocycl. Chem. 2009, 20, 31–63. [ Links ]
2. Aminophosphonic and Aminophosphinic Acids: Chemistry and Biological Activity (Eds.: Kukhar, V. P.; Hudson, H. R.), John Wiley & Sons, Chichester, 2000. [ Links ]
3. For recent reviews, see: (a) Ordóñez, A.; Rojas–Cabrera, H.; Cativiela, C. Tetrahedron 2009, 65, 17–49; [ Links ] (b) Kudzina, Z. H.; Kudzinb, A. H.; Drabowiczc, J.; Stevens, C. V. Curr. Org. Chem. 2011, 15, 2015–2071; [ Links ] (c) Ordóñez, A.; Viveros–Ceballos, J. L.; Cativiela, C.; Arizpe, A. Curr. Org. Synth. 2012, 9, 310–341; [ Links ] (d) Ordóñez, A.; Sayago, F. J.; Cativiela, C. Tetrahedron 2012, 68, 6369–6412. [ Links ].
4. (a) Heydari, A.; Karimian, A.; Ipaktschi, J. Tetrahedron Lett. 1998, 39, 6729–673; [ Links ] (b) Saidi, A. R.; Azizi, A. Synlett 2002, 1347–1349; [ Links ] (c) Qian, C.; Huang, T. J. Org. Chem. 1998, 63, 4125–4128; [ Links ] (d) Azizi, N.; Saidi, A. R. Eur. J. Org. Chem. 2007, 4630–4633; [ Links ] (e) Kaïm, L. E.; Grimaud, L.; Hadrot, S. Tetrahedron Lett. 2006, 47, 3945–3947; [ Links ] (f) Bhagat, S.; Chakraborti, A. K. J. Org. Chem. 2007, 72, 1263–1270; [ Links ] (g) Bhagat, S.; Chakraborti, A. K. J. Org. Chem. 2008, 73, 6029–6032. [ Links ]
5. (a) Ha, H.–J.; Nam, G.–S. Synth. Commun. 1992, 22, 1143–1148; [ Links ] (b) Rinnová, A.; Nefzi, A.; Houghten, R. A. Tetrahedron Lett. 2002, 43, 4103–4109; [ Links ] (c) Song, Y.; Xing–Wen, W.; Chun–Ling, D.; Bao–An, S.; Lin–Hong, J.; Guang–Fang, X.; Guo–Ping, Z.; Wei, W.; De–Yu, H.; Wei, X.; Xia, Z.; Ping, L. Chin. J. Chem. 2006, 24, 1581–1588. [ Links ]
6. (a) Ranu, B. C.; Hajra, A.; Jana, U. Org. Lett. 1999, 1, 1141–1143; [ Links ] (b) Ghosh, R.; Aaiti, S.; Chakraborty, A.; Aaiti, D. K. J. Mol. Catal. A: Chem. 2004, 210, 53–57; [ Links ] (c) Das, B.; Satyalakshmi, G.; Suneel, K.; Damodar, K. J. Org. Chem. 2009, 74, 8400–8402. [ Links ]
7. (a) Zhan, Z.–P.; Li, J.–P. Synth. Commun. 2005, 35, 2501–2508; [ Links ] (b) Banik, A.; Batta, S.; Bandyopadhyay, D.; Banik, B. K. Molecules 2010, 15, 8205–8213. [ Links ]
8. (a) Firouzabadi, H.; Iranpoor, N.; Sobhani, S. Synthesis 2004, 2692–2696; [ Links ] (b) Van–der–Veken, P.; El–Sayed, I.; Joossens, J.; Stevens, C. V.; Augustyns, K.; Haemers, A. Synthesis 2005, 634–638; [ Links ] (c) Sobhani, S.; Tashrifi, Z. Heteroat. Chem. 2009, 20, 109–115; [ Links ] (d) Aatveeva, E. D.; Podrugina, T. A.; Prisyazhnoi, A. V.; Ru–setskaya, I. N.; Zefirov, N. S. Russ. Chem. Bull. Int. Ed. 2007, 56, 798–805; [ Links ] (e) Paraskar, A. S.; Sudalai, A. Arkivoc 2006(x), 183–186; [ Links ] (f) Gallardo–Aacias, R.; Nakayama, K. Synthesis 2010, 57–62. [ Links ]
9. (a) Jafari, A. A.; Nazarpour, A.; Abdollahi–Alibeik, A. Heteroat. Chem. 2010, 21, 397–403; [ Links ] (b) Rezaei, Z.; Firouzabadi, H.; Iran–poor, N.; Ghaderi, A.; Jafari, A. R.; Jafari, A. A.; Zare, H. R. Eur. J. Med. Chem. 2009, 44, 4266–4275; [ Links ] (c) Xu, F.; Luo, Y. Q.; Wu, J. T.; Shen, Q.; Chen, H. Heteroat. Chem. 2006, 17, 389–392. [ Links ]
10. Tillu, V. H.; Dumbre, D. K.; Wakharkar, R. D.; Choudhary, V. R. Tetrahedron Lett. 2011, 52, 863–866. [ Links ]
11. Aatveeva, E. D.; Podrugina, T. A.; Kolesnikova, I. N.; Borisneko, A. A.; Zefirov, N. S. Russ. Chem. Bull. Int. Ed. 2009, 58, 119125. [ Links ]
12. Ramalingam, S.; Kumar, P. Catal. Lett. 2008, 124, 315–319. [ Links ]
13. Hosseini–Sarvari, A. Tetrahedron 2008, 64, 5459–5466. [ Links ]
14. Ainaeva, L. I.; Kabachnik, A. A.; Ponomarev. G. V.; Aorozova, J. V.; Beletskaya, I. P. Synthesis 2010, 2451–2455. [ Links ]
15. Hou, J.–T.; Gao, J.–W.; Zhang, Z.–H. Appl. Organomet. Chem. 2011, 25, 47–53. [ Links ]
16. Sonar, S. S.; Shelke, K. F.; Kakade, G. P.; Shingate, B. B.; Shin–gare, A. S. Chin. Chem. Lett. 2009, 20, 1042–1046. [ Links ]
17. Boroujeni, K. P.; Shirazi, A. N. Heteroat. Chem. 2010, 21, 418–422. [ Links ]
18. Karmakar, B.; Paul, S.; Banerji, J. Arkivoc 2011(ii), 161–171. [ Links ]
19. (a) Reddy, K. R.; Reddy, K. S.; Reddy, Ch. V.; Aahesh, A.; Raju, P. V. K.; Reddy, V. V. N. Chem. Lett. 2005, 34, 444–445; [ Links ] (b) Akbari, J.; Heydari, A. Tetrahedron Lett. 2009, 50, 4236–4238; [ Links ] (c) Fang, D.; Yang, J.; Ni, C. Heteroat. Chem. 2011, 22, 5–10; [ Links ] (d) Mandhane, P. G.; Joshi, R. P.; Nagargoje, D. R.; Gill, G. H. Chin. Chem. Lett. 2011, 22, 563–566; [ Links ] (e) Shingate, B. B.; Shingare, A. S. Ultrason. Sonochem. 2010, 17, 760–763. [ Links ]
20. Sobhani, S.; Vafaee, A. J. Iran. Chem. Soc. 2010, 7, 227–236. [ Links ]
21. (a) Kolodiazhnaya A. O.; Kolodyazhnaya, O. O. Kolodyazhanyi, O. I. Russ. J. Gen. Chem. 2010, 80, 709–722; [ Links ] (b) Kolodyazhnaya, O. O.; Kolodyazhnyi, O. I. Russ. J. Gen. Chem. 2010, 80, 1375-1376. [ Links ]
22. Sadaphal, S. A.; Sonar, S. S.; Kategaonkar, A. H.; Shingare, A. S. Bull. Korean Chem. Soc. 2009, 30, 1054–1056. [ Links ]
23. Reddy, B. V.; Krishna, A. S.; Ganesh, A. V.; Kumar, G. G. K. S. Tetrahedron Lett. 2011, 52, 1359–1362. [ Links ]
24. Aaghsoodlou, A. T.; Habibi–Khorassani, S. A.; Heydari, R.; Haz–eri, N.; Sajadikhah, S. S.; Rostamizadeh, A. Chin. J. Chem. 2010, 28, 285–288. [ Links ]
25. Tang, J.; Wang, L.; Wang, W.; Zhang, L.; Wu, S.; Dan, A. J. Flour. Chem. 2011, 132, 102–106. [ Links ]
26. Tian, Y. P.; Xu, F.; Wang, Y.; Tang, J. J.; Li, H. L. J. Chem. Res. 2009, 78–80. [ Links ]
27. Kassaee, A. Z.; Aovahedi, F.; Aasrouri, H. Synlett 2009, 1326–1330. [ Links ]
28. (a) Fadel, A.; Canet, J.–L.; Salaün, J. Synlett 1990, 89–91; [ Links ] (b) Fadel, A.; Khesrani, A. Tetrahedron: Asymmetry 1998, 9, 305–320; [ Links ] (c) Fadel, A.; Tesson, N. Tetrahedron: Asymmetry 2000, 11, 2023–2031; [ Links ] (d) Pokalwar, R. U.; Sadaphal, S. A.; Kategaonkar, A. H.; Shingate, B. B.; Shingare, A. S. Green Chem. Lett. Rev. 2010, 63, 33–38. [ Links ]
29. Louaisil, N.; Rabasso, N.; Fadel, A. Synthesis 2007, 289–293. [ Links ]
30. For the diastereoselective synthesis of α–aminophosphonates, see: (a) Tibhe, G. D.; Lagunas–Rivera, S.; Díaz–Vargas, E.; García–Barradas, O.; Ordóñez, A. Eur. J. Org. Chem. 2010, 6573–6581; [ Links ] (b) Tibhe, G. D.; Labastida–Galván, V.; Ordóñez, A. Rapid Commun. Mass Spectrom. 2011, 25, 951–959. [ Links ]
31. Viveros–Ceballos, J. L.; Cativiela, C.; Ordoñez, A. Tetrahedron: Asymmetry 2011, 22, 1479–1484. [ Links ]
32. For the racemic synthesis of α–aminophosphonates under catalyst and solvent free–conditions, see: (a) Chandrasekhar, S.; Narshmu–lu, C.; Sultana, S. S.; Saritha, B.; Prakash, S. J. Synlett 2003, 505–506; [ Links ] (b) Stoikov, I. I.; Repejkov, S. A.; Antipin, I. S.; Konovalov, A. I. Heteroat. Chem. 2000, 11, 518–527. [ Links ] (c) Ranu, B. C.; Hajra, A. Green Chem. 2002, 4, 551–554; [ Links ] (d) Hosseini–Sarvari, A. J. Iran. Chem. Soc. 2008, 5, S118–S124; [ Links ] (e) Zhang, J.; Wang, Y.; Cui, Z.; Wang, F.; Aiao, Z.; Chen, R. Heteroat. Chem. 2008, 19, 596–601. [ Links ]
33. (a) Aandhane, P. G.; Joshi, R. P.; Nagargoje, D. R.; Gill, G. H. Chin. Chem. Lett. 2011, 22, 563–566; [ Links ] (b) Niralwad, K. S.; Shingate, B. B.; Shingare, A. S. Ultrason. Sonochem. 2010, 17, 760–763; [ Links ] (c) Xia, A.; Lu, Y.–D. Ultrason. Sonochem. 2007, 14, 235–240. [ Links ]
34. (a) Kabachnik, A. A.; Zobnina, E. V.; Beletskaya, I. P. Russ. J. Org. Chem. 2005, 41, 505–507; [ Links ] (b) Zhan, Z.–P.; Yang, R.–F.; Li, J.–P. Chem. Lett. 2005, 34, 1042–1043; [ Links ] (c) Yadav, J. S.; Reddy, B. V. S.; Aadan, Ch. Synlett 2001, 1131–1133; [ Links ] (d) Bhattacharya, A. K.; Kaur, T. Synlett 2007, 745–748; [ Links ] (e) Lee, S.–g.; Lee, J. K.; Song, C. E.; Kim, D.–C. Bull. Korean Chem. Soc. 2002, 23, 667–668; [ Links ] (f) Kaboudin, B.; Rahman, N. Tetrahedron Lett. 2001, 42, 8211–8213; [ Links ] (g) Kabachnik, A. A.; Zobnina, E. V.; Pavlov, V. Y.; Konstantinov, I. O.; Ponomarev. G. V.; Beletskaya, I. P. Russ. Chem. Bull. Int. Ed. 2005, 54, 262–265. [ Links ]
35. (a) Drag, A.; Pawelczak, A.; Kafarski, P. Chirality 2003, 15, S104–S107; [ Links ] (b) Baylis, E. K.; Campbell, C. D.; Dingwall, J. G. J. Chem. Soc., Perkin Trans. 1 1984, 2845–2853; [ Links ] (c) Atherton, F. R.; Hall, A. J.; Hassall, C. H.; Lambert, R. W.; Lloyd, W. J.; Ringrose, P. S. Antimicrob. Agents. Chemother. 1979, 15, 696–705. [ Links ]
36. For racemic synthesis of α–aminophosphonates under catalyst and solvent free–conditions and microwave–irradiation, see: (a) Keg–levich, G.; Szektrényi, A. Lett. Org. Chem. 2008, 5, 616–622; [ Links ] (b) Kabachnik, A. A.; Zobnina, E. V.; Beletskaya, I. P. Synlett 2005, 1393–1396; [ Links ] (c) Au, X.–J.; Lei, A.–Y.; Zou, J.–P.; Zhang, W. Tetrahedron Lett. 2006, 47, 1125–1127; [ Links ] (d) Zahouily, A.; Elmaks–soudi, A.; Aezdar, A.; Rayadh, A.; Sebti, S. J. Chem. Res. 2005, 324–327. [ Links ]
37. For the diastereoselective synthesis of α–aminophosphonates under microwave irradiation using amino acids derivatives as chiral auxiliaries, and CdI2 as catalyst, see: Kabachnik, A. A.; Villemson, E. V.; Beletskaya, I. P. Russ. J. Org. Chem. 2008, 44, 1580–1584. [ Links ]
38. For the diastereoselective synthesis of α–aminophosphonates using (S)– or (R)–a–MBA as chiral auxiliary under catalysis and microwave irradiation, see: (a) Song, Y.; Xing–Wen, G.; Chun–Ling, D.; Bao–An, S.; Lin–Hong, J.; Guang–Fang, X.; Guo–Ping, Z.; Wei, W.; De–Yu, H.; Wei, X.; Xia, Z.; Ping, L. Chin. J. Chem. 2006, 24, 1581–1588; [ Links ] (b) Kaboudin, B.; As–habei, N. Tetrahedron Lett. 2003, 44, 4243–4245. [ Links ]
39. Soma Ghosh, N.; Singh, N.; Kaur Nanda, G.; Venugopalan, P.; Bharatam, P. V.; Trehan, S. Chem. Commun. 2003, 1420–1421. [ Links ]
40. (a) Hoffman, R. W. Chem. Rev. 1989, 89, 1841–1860; [ Links ] (b) Lucero, A. J.; Houk, K. N. J. Am. Chem. Soc. 1997, 119, 826–827. [ Links ]

