










Services on Demand
Journal

Article
 English (pdf)
English (pdf)
 Article in xml format
Article in xml format Article references
Article references
 Send this article by e-mail
Send this article by e-mailIndicators
 Cited by SciELO
Cited by SciELO Related links
 Similars in
SciELO
Similars in
SciELO Share

 Permalink
PermalinkJournal of the Mexican Chemical Society
Print version ISSN 1870-249X
J. Mex. Chem. Soc vol.56 n.2 Ciudad de México Apr./Jun. 2012
Article
Infrared Irradiation–Assisted Multicomponent Synthesis of 2–Amino–3–cyano–4H–pyran Derivatives
Arturo Sánchez,1 Fernando Hernández,2 Paulo César Cruz,1 Yolanda Alcaraz,3 Joaquín Tamariz,1 Francisco Delgado,1 and Miguel A. Vázquez2*
1 Departamento de Química Orgánica, Escuela Nacional de Ciencias Biológicas–IPN, Prolongación Carpio y Plan de Ayala S/N, 11340 México, D. F., México. *mvazquez@ugto.mx
2 Departamento de Química, Universidad de Guanajuato, Noria Alta S/N, 36050 Guanajuato, Gto., México.
3 Departamento de Farmacia, Universidad de Guanajuato, Noria Alta S/N, 36050 Guanajuato, Gto., México.
Received December 12, 2011.
accepted February 3, 2012.
Abstract
A simple, versatile, and efficient synthesis of 4H–pyran derivative compounds is achieved via a three–component cyclocon–densation of aldehyles, malononitrile, and ethyl acetoacetate, using ammonium hydroxide as the catalyst, promoted by infrared irradiation. The present method offers several advantages, such as high yields, non hazarlous reaction conditions as well as short reaction times.
Key words: Infrared Irradiation, three–component reaction, MCR, 4H–pyrans.
Resumen
Se informa una síntesis sencilla, versátil y eficiente de derivados de 4H–piranos, a través de una ciclocondensación de tres componentes entre aldehílos, malononitrilo y acetoacetato de etilo, empleando hidróxido de amonio como catalizador y promovida por irradiación infrarroja como fuente de activación alterna. Este método ofrece varias ventajas, tal como: elevados rendimientos, condiciones no peligrosas y cortos tiempos de reacción.
Palabras clave: Irradiación infrarroja, reacción de tres componentes, RMC, 4H–piranos.
Introduction
Multicomponent reactions (MCRs) have greatly contributed to the convergent synthesis of complex and structurally interesting organic molecules from simple and realily available starting materials, and have emerged as a powerful tool for drug discovery [1]. In the other hand, 4H–pyrans are an important class of heterocycles because the core fragment is constituted by a great variety of natural products and biologically active compounds. The latter compounds, which have many pharmacological properties and play important roles in biochemical processes [2], include a selective inhibitor of excitatory amino acil transporter subtype 1 [3], IKCa channel blockers [4], A3 adenosine receptor antagonists as potential anti–inflammatory agents [5], and pro–apoptotic activity against cancer cells (administered alone or in combination with chemo– and radio–therapy) [6]. In addition, they can be used as cognitive enhancers for the treatment of neurodegenerative diseases [7]. Therefore, the synthesis of 4H–pyrans and their derivatives has attracted much attention in organic synthesis [8].
The typical procedure to synthesize pyran derivatives is usually a two–step reaction carried out between a Michael acceptor (arylidenemalononitriles) and β–dicarbonyl compounds in the presence of a base as the catalyst (piperidine, morpholine or metal alkoxides) in ethanol or other solvents (DMSO, DMF). A variety of methods have been used to prepare 4H–pyrans, which involve several catalysts such as KF/Al2O3 [8c], Baker's yeast [8e], hexadecyldimethylbenzyl ammonium bromide (HD–MBAB) [9], InBr3 [10], and H6P2W18O6218H2O [11]. In addition to the employment of alternative activating sources, such as microwave irradiation [12, 13], combined microwave and ultrasound irradiation [14], and electro–oxidation [15], sometimes an ionic liquid is employed [12]. Although these methods are valuable, they suffer from one or more disadvantages, such as hazardous reagents, high temperature, long reaction time, low yields, and tedious workup. Hence, it is important to continue to search for simpler and more efficient protocols.
One of the objectives in modern synthetic organic chemistry is to carry out reactions with effective, clean, economical, and environmentally safer methodologies [16]. In this sense, the synthetic reactions using infrared irradiation, as a source of heating, constitutes a significant advance in the development of environmentally benign methods [17], being one of the main focuses of our research group. In this context, numerous reactions have been included in our studies, among them the Knoevenagel condensation [18], the Biginelli reaction [19], the direct conversion of aromatic aldehydes into the corresponding nitriles [20], the Fischer indole reaction [21], the formation of N–benzylideneanilines [22], and more recently, the molecular rearrangement of perezone into isoperezone [23].
In keeping with our interest in the application of infrared irradiation in organic synthesis, we herein disclose a simple and efficient synthesis of 4H–pyran derivatives via a three–component cyclocondensation reaction of aldehydes, malononitrile, and ethyl acetoacetate using ammonium hydroxile as the catalyst. To the best of our knowledge, this is the first time that this cyclocondensation has been promoted by infrared irradiation.
Results and discussion
Initially, we carried out the cyclocondensation reaction of benzaldehyle (1a), malononitrile (2), ethyl acetoacetate (3) and commercial ammonium hydroxile (1:1:1 equiv. mol and 28 mol%, respectively) at reflux temperature for 1 h, which led to a poor yield (45%) of 4H–pyran 4a (Table 1, entry 1). In spite of increasing the reaction time to 2 h, no appreciable increment was observed in the yield of the desired product.
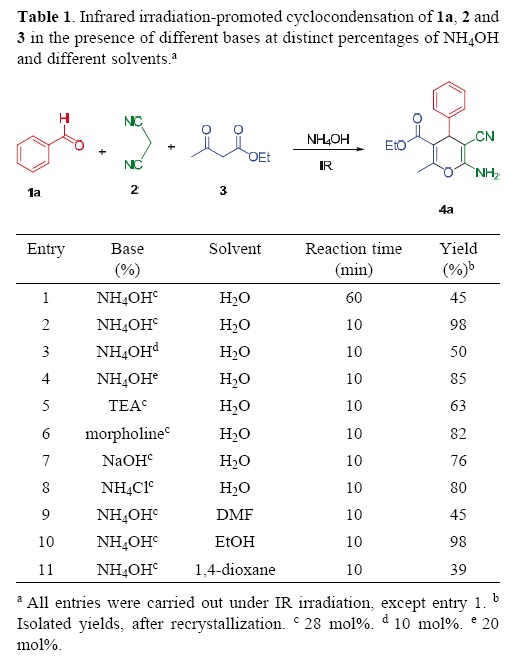
Therefore, an attempt was male to promote the target reaction by employing infrared irradiation as the source of heating, by taking into account that we have previously demonstrated that Knoevenagel adducts can be easily prepared by using infrared irradiation for the condensation of benzaldehyles and malononitrile in solvent–free conditions [18]. Interestingly, when this protocol was applied to the condensation of benzaldehyle (1a), malononitrile (2), ethyl acetoacetate (3), and commercial ammonium hydroxile, the 4H–pyran derivative 4a (Table 1, entry 2) was isolated in excellent yield (98%) after only 10 min of reaction.
Thus, we evaluated the amount of ammonium hydroxide required for this transformation. By using 10, 20 and 40 mol% of the base under same reaction conditions, we obtained 50%, 85% and 97% yields, respectively. The highest yield was obtained (98%) when the reaction was loaded with 28 mol% of the base (Table 1, entries 2–4). Increasing this catalyst beyond that concentration did not affect the yield. It is important to note that in order to obtain the product 4a in pure form, a simple filtration and recrystallization were carried out.
A series of organic and inorganic bases were then tested and compared to ammonium hydroxile. As shown in Table 1 (entries 5–8), good yields were obtained with triethylamine, morpholine, sodium hydroxide or ammonium chloride as abase. However, afterwards the purification of the desirel product was more difficult in comparison with the use of amonium hydroxile.
Finally, we examined the effect of the solvent on the reaction. When using protic and aprotic solvents, EtOH afforded the product in excellent yield (98%), whereas DMF and dioxane gave only moderate yields (Table 1, entries 9, 10 and 11).
According to these results, we decided to perform the reaction with the mixture of 1a, 2, and 3 (1:1:1 equiv. mol), and NH4OH (28 mol%). This mixture was irradiated with an infrared lamp (OSRAM R–20 bulb, 127 V, 250 W, λ = 1255.6 nm) [24, 25, 26] at reflux temperature for 10 min, under solvent–free conditions (Table 1, entry 2). After this time, the conversion of the substrates into 4a was very efficient, leading to the desired 4H–pyran 4a in high yield (98%).
To validate this new method and to assess the effect of different substituents on the formation of the 4H–pyrans, both electron–poor anl electron–rich aromatic aldehydes, as well as heteroaromatic and aliphatic aldehyles were employed. In all cases, almost the same result of cyclocondensation was observed, giving the desired adduct (Table 2). Aliphatic aldehyles gave the corresponding 4H–pyrans in lower yield (45–58%) (entries 12–13) than aromatic and heterocyclic aldehyles. However, no obvious correlation was found between the nature of the substituent on the aromatic or heterocyclic ring and the yield of this conversion. One of the major advantages of this protocol is the isolation and purification of the products, which have been achieved by simple washing and crystallization of the crude products.

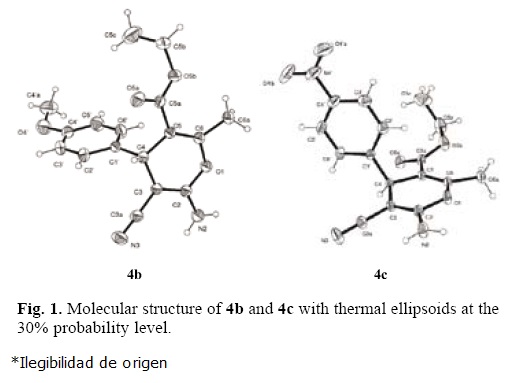
The structural elucidation of products 4a–m was made on the basis of their spectroscopic data. Thus, for instance, infrared (IR) spectra of 4c exhibited bands at 3335 (NH2), 2194 (CN), and 1676 (C=O) cm–1. The 1H NMR spectrum of 4c showed the presence of: (i) four aromatic protons at 7.42–8.16 ppm attributed to system AA'–BB'; (ii) a triplet and quartet at 1.01 and 3.97 ppm integrating for three and two protons of the ethoxycarbonyl group, respectively; and (iii) three singlet at 2.38, 4.47 and 6.86 ppm identified as the methine (C–4), methyl (CH3), and NH2 protons. The 13C NMR spectrum displayed: (i) a signal at 169.1 ppm due to the carbonyl group, and at 117.4 ppm due to cyano; (ii) four signals for the vinyl carbons at 158.2 (C–2), 156.1 (C–6), 104.3 (C–5), and 54.7 ppm (C–3); and (iii) four signal for aromatic carbons at 150.6–121.7 ppm. The attributions of the signals were supported on 2D experiments such as HMQC and HMBC. A single–crystal determination for compounds 4b and 4c allowed us to confirm their molecular structure (Figure 1). The X–ray structures show that the aryl group in C–4 and the heterocyclic ring are almost orthogonal. A torsion angle C(3)–C(4)–C(1')–C(2') of 91.38° (18) was found for 4b and another C(3)–C(4)–C(1')–C(6') of –96.99° (15) for 4c. [27]
The formation of 4 can be explained by the possible mechanism presented in Scheme 1. The reaction can occur via the initial formation of the α,ß–unsaturated compound A, from the Knoevenagel condensation reaction between aldehyle 1 and malononitrile (2), which undergoes nucleophilic attack of the anion of ethyl acetoacetate (3) to give the Michael adduct B. Then, the latter promotes the cyclization to intermediate C, and subsequent tautomerization to afford the polyfunctional–ized 4H–pyrans 4. However, an alternative mechanism cannot be ruled out, in which the pathway starts from the reaction between 1 and 3 to generate the α,ß–unsaturated compound E, which in turn undergoes the conjugated addition of the anion of malononitrile to afford adduct B. This latter possible mechanism follows the same sequence of reactions shown in the first case, giving rise to adduct 4.
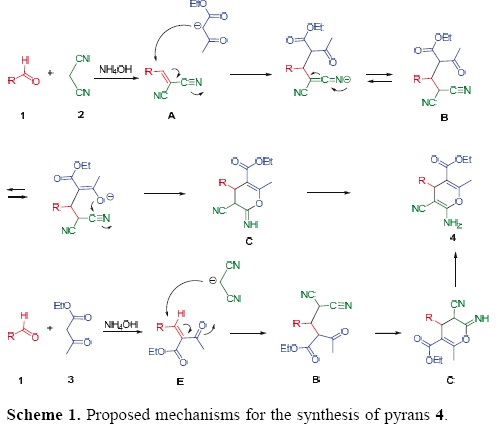
To test of the proposed mechanism, the reaction between benzylidenemalononitriles 5a–d, ethyl acetoacetate (3) and NH4OH (28 mol%) was carried out under infrared irradiation. The target compounds 4a–c and 4h were obtained in slightly lower yields than those obtained by the three–component reaction (Table 3). In contrast, when we attempted to test the second mechanistic route, starting from benzaldehyle (1a) and ethyl acetoacetate (3), the product of the Knoevenagel reaction, E, was not formed, leading to a complex mixture of products.

Conclusion
A novel and efficient three–component method for the synthesis of 4H–pyran derivatives, catalyzed with NH4OH and promoted by infrared irradiation is described. Compared with other procedures, this method has the advantage of being a single step and easy operation with short reaction times, mild and non–hazardous (environmentally friendly) reaction conditions, and good yields of potentially active compounds. Moreover, this method further expands the application of a new and efficient source of energy, infrared irradiation, in organic synthesis.
Experimental section
General Procedure. Melting points were determined on a Fisher–Johns melting point apparatus and were uncorrected. The progress of the reaction and the purity of compounds were monitored by TLC with E. Merck silica gel 60–F254 coated aluminum sheets, in w–hexane/ethyl acetate (7:3), and visualized by a 254 nm UV lamp. IR spectra were recorded on a Perkin–Elmer 2000 spectrophotometer. NMR spectra were recorded, for solutions in DMSO–d6 and CDCl3 with Me4Si as internal standard, on Varian Gemini (200 MHz) and Varian VNMR System (500 MHz) instruments. High–resolution mass spectra (HRMS) were obtained with a JSM–GCMate II mass spectrometer, and electron impact techniques (70 eV) were employed. X–ray diffraction data were collected on an Oxford Diffraction Xcalibur S single–crystal X–ray diffractometer.
General procedures for the preparation of 2–amino–3–cyano–4/f–pyrans derivatives (4a–m). Method A. A mixture of aldehyle 1a–m (3.0 mmol), malononitrile 2 (3.0 mmol), ethyl acetoacetate 2 (3.0 mmol) and ammonium hydroxide solution 28% w/w (373 mg, 3.0 mmol) was placed in a 25 mL round–bottom flask equipped with a condenser. The mixture was irradiated with an infrared lamp at reflux temperature for –10 min, under solvent–free conditions, until the consumption of the substrates. The reaction was monitored by 1H NMR or tlc. After cooling, the product mixture was directly submitted to recrystallization from water/ethanol (90:10), affording the corresponding pure 4H–pyran derivates 4a–m.
Method B. A mixture of the benzylidenemalononitriles 5a–d (1 mol–equiv.), ethyl acetoacetate 3 (1 mol–equiv.) and an aqueous solution of ammonium hydroxide, 28% w/w (373 mg, 3.0 mmol) was placed in a 25 mL round–bottom flask equipped with a condenser. The mixture was irradiated with an infrared lamp [24] for –5–30 min, under solvent–free conditions, until the consumption of the substrates. After cooling, the product mixture was directly submitted to recrystallization from water/ ethanol (90:10), affording the corresponding pure 4H–pyran derivates 4a–c and 4h.
The target compounds were fully characterized by their physical and spectral data. In the case of known compounds, their physical and spectroscopic data were compared with those reported in the literature and found to be related. 4a, mp 195–196 °C (lit. [28a]), 4b mp 142–144 °C 4c, mp 182–183 °C (lit.[28a]), (lit.[28c]), 4d, mp 178–179 °C (lit.[28a], 4f, mp 225–227 °C (lit. [28c]), 4g, mp 153–156 °C (lit. [28a]),), 4h, mp 154–157 °C (lit. [28a]), 4i, mp 162–163 °C (lit. [28c]), 4j, mp 280 °C (lit. [28c]), 4k, mp 135–137 °C (lit. [28c]).
2–Amino–3–cyano–5–ethoxycarbonyl–6–methyl–4–phe–nyl–4//–pyran (4a). Following General Method A, the reaction between 1a (319 mg, 3.0 mmol), malononitrile (2) (198 mg, 3.0 mmol), ethyl acetoacetate (3) (390 mg, 3.0 mmol) and ammonium hydroxide 28% w/w (373 mg, 3.0 mmol) was irradiated with infrared irradiation for 5 min. The crude product was recrystallized from water/ethanol, giving 766 mg (90%) of 4a as a white solid; mp 182–184 °C.
Method B. A mixture of benzylidenemalononitrile 5a (462 mg, 3.0 mmol), ethyl acetoacetate (3) (390 mg, 3.0 mmol) and ammonium hydroxide 28% w/w (373 mg, 3.0 mmol) was irradiated with infrared irradiation for 15 min. The crude product was recrystallized from water/ethanol, giving 596 mg (70%) of 4a; FT–IR (KBr) vmax 3404, 2190, 1688 cm–1; 1H NMR (200 MHz, DMSO–d6) δ 1.03 (t, J = 7.0 Hz, 3H, OCH2CH3), 2.31 (s, 3H, CH3), 3.87 (qd, J = 7.0, 1.4 Hz, 2H, OCH2CH3), 4.29 (s, 1H, H–4), 6.78 (br s, 2H, NH2), 7.12–7.31 (m, 5H, H–Ar); 13C (50 MHz, DMSO–d6) δ 13.6 (OCH2CH3), 18.1 (CH3), 38.2 (C–4), 57.3 (C–3), 60.0 (OCH2CH3), 107.2 (C–5), 119.6 (C≡N), 126.6 (C–4'), 127.1 (C–2'and C–6'), 128.2 (C–3'and C–5'), 144.8 (C–1'), 156.5 (C–6), 158.3 (C–2), 165.3 (C=O). HRMS (EI+) calcd for C16H16N2O3 284.1161, found (M+ ) 284.1158.
2–Amino–3–cyano–5–ethoxycarbonyl–4–(4–methoxyphenyl)–6–methyl–4H–pyran (4b). Following General Method A, the reaction between 1b (408 mg, 3.0 mmol), malononitrile (2) (198 mg, 3.0 mmol), ethyl acetoacetate (3) (390 mg, 3.0 mmol) and ammonium hydroxide 28% w/w (373 mg, 3.0 mmol) was irradiated with infrared irradiation for 10 min. The crude product was recrystallized from water/ethanol, giving 753 mg (80%) of 4b as a yellow solid; yield; mp 135–137 °C.
Method B. A mixture of benzylidenemalononitrile 5b (552 mg, 3.0 mmol), ethyl acetoacetate (3) (390 mg, 3.0 mmol) and ammonium hydroxide 28% w/w (373 mg, 3.0 mmol) was irradiated with infrared irradiation for 35 min. The crude product was recrystallized from water/ethanol, giving 642 mg (68%) of 4b; IR (KBr) vmax 3400, 2191, 1682 cm–1; 1H NMR (200 MHz, CDCl3) δ 1.11 (t, J = 7.1 Hz, 3H, OCH2CH3), 2.34 (s, 3H, CH3), 3.77 (s, 3H, OCH3), 4.02 (q, J = 7.2 Hz, 2H, OCH2CH3), 4.39 (s, 1H, H–4), 4.56 (br s, 2H, NH2), 6.82 (d, J = 8.6 Hz, 2H, H–3' and H–5'), 7.12 (d, J = 8.6 Hz, 2H, H–2' and H–6'); 13C (50 MHz, DMSO–d6) δ 13.9 (OCH2CH3), 18.3 (CH3), 37.9 (C–4), 55.2 (OCH3), 60.6 (OCH2CH3), 62.3 (C–3), 108.19 (C5), 113.9 (C–3' and C–5'), 119.1 (C≡N), 128.6 (C–2' and C–6'), 136.1 (C–1'), 156.3 (C–4'), 157.4 (C–6), 158.6 (C– 2), 160.1 (C=O). HRMS (EI+) calcd for C17H18N2O4 314.1267, found (M+) 314.1268.
2–Amino–3–cyano–5–ethoxycarbonyl–6–methyl–4–(4–ni–trophenyl)–4H–pyran (4c). Following General Method A,the reaction between 1c (453 mg, 3.0 mmol), malononitrile (2) (198 mg, 3.0 mmol), ethyl acetoacetate (3) (390 mg, 3.0 mmol) and ammonium hydroxide 28% w/w (373 mg, 3.0 mmol) was irradiated with infrared irradiation for 5 min. The crude product was recrystallized from water/ethanol, giving 967 mg (98%) of 4c as a yellow solid; yield; mp 178–180 °C.
Method B. A mixture of benzylidenemalononitrile 5c (597 mg, 3.0 mmol), ethyl acetoacetate (3) (390 mg, 3.0 mmol) and ammonium hydroxide 28% w/w (373 mg, 3.0 mmol) was irradiated with infrared irradiation for 10 min. The crude product was recrystallized from H2O, giving 888 mg (90%) of 4c; IR (KBr) vmax 3335, 2194, 1676 cm–1; 1H NMR (200 MHz DMSO–d6) δ 1.01 (t, J = 7.0 Hz, 3H, OCH2CH3), 2.38 (s, 3H, CH3), 3.97 (q, J = 7.0 Hz, 2H, OCH2CH3), 4.47 (s, 1H, H–4), 6.86 (br s, 2H, NH2), 7.42 (d, J = 8.8 Hz, 2H, H–2' and H–6'), 8.16 (d, J = 8.8 Hz, 2H, H–3'and H–5'); 13C (50 MHz DMSO–d6) δ 11.95 (OCH2CH3), 16.6 (CH3), 36.6 (C–4), 54.7 (C–3), 58.4 (OCH2CH3), 104.3 (C–5), 117.4 (C≡N), 121.7 (C–2' and C–6'), 126.5 (C–3' and C–5'), 146.6 (C–4'), 150.6 (C–1'), 156.1 (C–6), 158.2 (C–2), 169.1 (C=O). HRMS (EI+) calcd for C16H15N3O5 329.1012, found (M+) 329.1025.
2–Amino–3–cyano–5–ethoxycarbonyl–6–methyl–4–(3–ni–trophenyl)–4H–pyran (4d). Following General Method A, the reaction between 1d (453 mg, 3.0 mmol), malononitrile (2) (198 mg, 3.0 mmol), ethyl acetoacetate (3) (390 mg, 3.0 mmol) and ammonium hydroxide 28% w/w (373 mg, 3.0 mmol) was irradiated with infrared irradiation for 5 min. The crude product was recrystallized from water/ethanol, giving 868 mg (88%) of 4d as a yellow solid; yield; mp 179–181 °C; IR (KBr) vmax 3455, 2209, 1689 cm–1; 1H NMR (200 MHz, DMSO–d6) δ 0.90 (t, J = 7.2 Hz, 3H, OCH2CH3), 2.33 (s, 3H, CH3), 3.85 (q, J = 7.2 Hz, 2H, OCH2CH3), 5.01 (s, 1H, H–4), 7.09 (br s, 2H, NH2), 7.45 (m, 2H, H–4' and H–6'), 7.69 (t, J = 8.0 Hz, 1H, H–5'), 7.84 (d, J = 8.0 Hz, 1H, H–3'); 13C (50 MHz, DMSO–d6) δ 13.1 (OCH2CH3), 17.9 (CH3), 32.5 (C–4), 55.5 (C–3), 58.9 (OCH2CH3), 105.9 (C–5), 118.5 (C≡N), 123.3 (C–3'), 127.6 (C–4'), 130.0 (C–2'), 133.3 (C–3'), 139.1 (C–1'), 148.0 (C–6'), 157.8 (C–6), 158.5 (C–2), 164.8 (C=O). HRMS (EI+) calcd for C16H15N3O5 329.1012, found (M+) 329.1013.
1,4–bis–(2–Amino–3–cyano–5–ethoxycarbonyl–6–methyl–4H–pyran–4–yl)benzene (4e). Following General Method A, the reaction between 1e (402 mg, 3.0 mmol), malononitrile (2) (396 mg, 3.0 mmol), ethyl acetoacetate (3) (780 mg, 3.0 mmol) and ammonium hydroxide 28% w/w (373 mg, 3.0 mmol) was irradiated with infrared irradiation for 10 min. The crude product was recrystallized from water/ethanol, giving 1,352 mg (92%) of 4e as a yellow solid; yield; mp 1909–191 °C; IR (KBr) vmax 3396, 2192, 1689 cm–1; 1H NMR (300 MHz, DMSO–d6) δ 0.97 (t, J = 7.2 Hz, 6H, 2OCH2CH3), 2.30 (s, 6H, 2CH3), 3.95 (m, 4H, 2OCH2CH3), 4.25 (s, 2H, 2H–4), 6.90 (br s, 4H, 2NH2), 7.06 (s, 4H); 13C (75.4 MHz, DMSO–d6) δ 13.6 (OCH2CH3), 18.0 (CH3), 38.3 (C–4'), 57.0 (C–3'), 60.0 (OCH2CH3), 107.1 (C–5'), 119.7 (C≡N), 127.2 (C–2 and C3), 143.3 (C–1 and C–4), 156.6 (C–6'), 158.4 (C–2'), 165.3 (C=O). HRMS (EI+) calcd for C26H26N4O6 490.1852, found (M+) 490.1853.
1,3–bis–(2–Amino–3–cyano–5–ethoxycarbonyl–6–methyl–4H–pyran–4–yl)benzene (4f). Following General Method A, the reaction between 1f (402 mg, 3.0 mmol), malononitrile (2) (396 mg, 3.0 mmol), ethyl acetoacetate (3) (780 mg, 3.0 mmol) and ammonium hydroxide 28% w/w (373 mg, 3.0 mmol) was irradiated with infrared irradiation for 10 min. The crude product was recrystallized from water/ethanol, giving 588 mg (40%) of 4f as a yellow solid; yield; mp 166–167 °C; IR (KBr) vmax 3410, 2193, 1678 cm–1; 1H NMR (300 MHz, DMSO–d6) δ 1.02 (t, J = 7.2 Hz, 6H, 2OCH2CH3), 2.30 (s, 6H, 2CH3), 3.85 (m, 4H, 2OCH2CH3), 4.26 (s, 2H, 2H–4'), 6.89 (s, 1H, H–2) 6.92 (br s, 4H, 2NH2), 6.99 (dd, J = 7.8, 1.2 Hz, 2H, H–4 and H–6), 7.26 (t, J = 7.8 Hz, 1H, H–5); 13C (75.4 MHz, DMSO–d6) δ 13.6 (OCH2CH3), 18.0 (CH3), 38.5 (C–4'), 57.0 (C–3'), 60.0 (OCH2CH3), 107.1 (C–5'), 119.6 (C≡N), 125.5 (C–2), 125.6 (C–4 and C–6), 128.8 (C–5), 144.9 (C–1 and C–3), 156.6 (C–6'), 158.5 (C–2'), 165.3 (C=O). HRMS (EI+) calcd for C26H26N4O6 490.1852, found (M+) 490.1851.
2–Amino–4–(3–chlorophenyl)–3–cyano–5–ethoxycarbonyl–6–methyl–4H–pyran (4g). Following General Method A, the reaction between 1g (420 mg, 3.0 mmol), malononitrile (2) (198 mg, 3.0 mmol), ethyl acetoacetate (3) (390 mg, 3.0 mmol) and ammonium hydroxide 28% w/w (373 mg, 3.0 mmol) was irradiated with infrared irradiation for 10 min. The crude product was recrystallized from water/ethanol, giving 543 mg (57%) of 4g as a yellow solid; yield; mp 175–177 °C; IR (KBr) vmax 3331, 2193, 1676 cm–1; 1H NMR (200 MHz, DMSO–d6) δ 1.08 (t, J = 7.4 Hz, 3H, OCH2CH3), 2.37 (s, 3H, CH3), 4.02 (m, 2H, OCH2CH3), 4.38 (s, 1H, H–4), 7.06 (br s, 2H, NH2), 7.16 (m, 4H, H–2', H–4', H–5', H–6'); 13C (50 MHz, DMSO–d6) δ 13.6 (OCH2CH3), 18.2 (CH3), 38.5 (C–4), 56.6 (C–3), 60.2 (OCH2CH3), 106.5 (C–5), 119.5 (C≡N), 126.0 (C–2'), 126.8 (C–6'), 127.0 (C–3'), 130.4 (C–4'), 132.9 (C–5'), 147.4 (C–1), 157.2 (C–6), 158.5 (C–2), 165.2 (C=O). HRMS (EI+) calcd for C16H15N2O3Cl 318.0771, found (M+) 318.0774.
2–Amino–3–cyano–5–ethoxycarbonyl–4–(4–flourophenyl)–6–methyl–4H–pyran (4h). Following General Method A, the reaction between 1h (372 mg, 3.0 mmol), malononitrile (2) (198 mg, 3.0 mmol), ethyl acetoacetate (3) (390 mg, 3.0 mmol) and ammonium hydroxide 28% w/w (373 mg, 3.0 mmol) was irradiated with infrared irradiation for 10 min. The crude product was recrystallized from water/ethanol, giving 725 mg (80%) of 4h as a white solid; mp 170–172 °C.
Method B. A mixture of benzylidenemalononitrile 5d (516 mg, 3.0 mmol), ethyl acetoacetate (3) (390 mg, 3.0 mmol) and ammonium hydroxide 28% w/w (373 mg, 3.0 mmol) was irradiated with infrared irradiation for 20 min. The crude product was recrystallized from H2O, giving 688 mg (76%) of 4h; IR (KBr) vmax 3403, 2193, 1691 cm–1; 1H NMR (200 MHz, DMSO–d6) δ 1.11 (t, J = 7.0 Hz, 3H, OCH2CH3), 2.35 (s, 3H, CH3), 4.05 (q, J = 7.0 Hz, 2H, OCH2CH3), 4.39 (s, 1H, H–4), 5.89 (br s, 2H, NH2), 6.92–7.21 (AA'BB' system, 4H, H–2', H–3', H–5', and H–6'); 13C (50 MHz, DMSO–d6), δ 13.8 (OCH2CH3), 18.4 (CH3), 38.2 (C–4), 59.2 (C–3), 60.3 (OCH2CH3), 107.5 (C–5), 115.2 (J = 21.3 Hz, C–3' and C–5'), 119.5 (C≡N), 129.0 (J = 7.9 Hz, C–2' and C–6'), 140.3 (C–1), 158.3 (C–2), 161.0 (J = 243.5 Hz, C–4'), 165.8 (C=O). HRMS (EI+) calcd for C16H15N2O3F 302.1067, found 302.1069.
2–Amino–3–cyano–5–ethoxycarbonyl–6–methyl–4–(pyridin–4–yl)–4H–pyran (4i). Following General Method A, the reaction between 1i (322 mg, 3.0 mmol), malononitrile (2) (198 mg, 3.0 mmol), ethyl acetoacetate (3) (390 mg, 3.0 mmol) and ammonium hydroxide 28% w/w (373 mg, 3.0 mmol) was irradiated with infrared irradiation for 5 min. The crude product was recrystallized from water/ethanol, giving 735 mg (86%) of 4i as a white solid; mp 164–166 °C; IR (KBr) vmax 3327, 2192, 1675 cm–1; 1H NMR (200 MHz, DMSO–d6) δ 1.01 (t, J = 7.0 Hz, 3H, OCH2CH3), 2.35 (s, 3H, CH3), 3.94 (q, J = 7.0 Hz, 2H, OCH2CH3), 4.32 (s, 1H, H–4), 7.08 (br s, 2H, NH2), 7.18 (d, J = 5.8 Hz, 2H, H–2' and H–6'), 8.50 (d, J = 5.8 Hz, 2H, H–3' and H–5'); 13C (50 MHz, DMSO–d6) δ 13.6 (OCH2CH3), 18.2 (CH3), 38.3 (C–4), 52.9 (C–3), 60.3 (OCH2CH3), 105.0 (C–5), 119.3 (C≡N), 122.3 (C–2' and C–6'), 149.8 (C–3' and C–5'), 153.3 (C–1'), 158.0 (C–6), 158.6 (C–2), 165.1 (C=O). HRMS (EI+) calcd for C15H15N3O3 285.1113, found (M+) 285.1113.
2–Amino–3–cyano–5–ethoxycarbonyl–4–(furan–2–yl)–6–methyl–4H–pyran (4j). Following General Method A, the reaction between 1i (288 mg, 3.0 mmol), malononitrile (2) (198 mg, 3.0 mmol), ethyl acetoacetate (3) (390 mg, 3.0 mmol) and ammonium hydroxide 28% w/w (373 mg, 3.0 mmol) was irradiated with infrared irradiation for 5 min. The crude product was recrystallized from water/ethanol, giving 780 mg (95%) of 4i as an orange solid; mp 189–190 °C; IR (KBr) vmax 3329, 2192, 1687 cm–1; 1H NMR (200 MHz, DMSO–d6) δ 1.13 (t, J = 7.0 Hz, 3H, OCH2CH3), 2.29 (s, 3H, CH3), 4.07 (m, 2H, OCH2CH3), 4.46 (s, 1H, H–4), 6.07 (d, J = 3.1 Hz, 1H, H–4'), 6.35 (m, 1H, H–5'), 7.03 (br s, 2H, NH2), 7.52 (l, J = 1.6 Hz, 1H, H–3'); 13C (50 MHz DMSO–d6) δ 13.8 (OCH2CH3), 18.2 (CH3), 32.4 (C–4), 54.1 (C–3), 60.3 (OCH2CH3), 105.0 (C–5'), 105.3 (C–5), 110.4 (C–4'), 119.5 (C≡N), 142.1 (C–3'), 155.9 (C–1'), 157.6 (C–6), 159.5 (C–2), 165.2 (C=O). HRMS (EI+) calcd for C14H14N2O4 274.0954, found (M+) 274.0956.
2–Amino–3–cyano–5–ethoxycarbonyl–4–(thiophen–2–yl)–6–methyl–4H–pyran (4k). Following General Method A, the reaction between 1k (335 mg, 3.0 mmol), malononitrile (2) (198 mg, 3.0 mmol), ethyl acetoacetate (3) (390 mg, 3.0 mmol) and ammonium hydroxide 28% w/w (373 mg, 3.0 mmol) was irradiated with infrared irradiation for 10 min. The crude product was recrystallized from water/ethanol, giving 696 mg (80%) of 4k as a pale yellow solid; mp 180–182; IR (KBr) vmax 3394, 2192, 1669 cm–1; 1H NMR (200 MHz, DMSO–d6) δ 1.14 (t, J = 7.2 Hz, 3H, OCH2CH3), 2.28 (s, 3H, CH3), 4.08 (dq, J = 7.2, 1.4 Hz, 2H, OCH2CH3), 4.61 (s, 1H, H–4), 6.82 (dd, J = 7.2, 1.4 Hz 1H, H–5'), 6.91 (m, 1H, H–4'), 7.05 (br s, 2H, NH2), 7.36 (dd, J = 7.2, 1.4 Hz, 1H, H–3'); 13C (50 MHz DMSO–d6) δ 13.8 (OCH2CH3), 18.1 (CH3), 33.8 (C–4), 56.9 (C–3), 60.4 (OCH2CH3), 107.6 (C–5), 119.5 (C≡N), 124.0 (C–5'), 124.7 (C–3'), 126.8 (C–4'), 149.3 (C–1'), 156.7 (C–6), 159.05 (C–2), 165.2 (C=O). HRMS (EI+) calcd for C14H14N2O3S 290.0725, found (M+) 290.0724.
2–Amino–3–cyano–5–ethoxycarbonyl–6–methyl–4H–pyran (4l). Following General Method A, the reaction between 1l (90 mg, 3.0 mmol), malononitrile (2) (198 mg, 3.0 mmol), ethyl acetoacetate (3) (390 mg, 3.0 mmol) and ammonium hydroxide 28% w/w (373 mg, 3.0 mmol) was irradiated with infrared irradiation for 10 min. The crude product was recrystallized from water/ethanol, giving 280 mg (45%) of 4l as a pale yellow solid; mp 252–254 °C; IR (KBr) vmax 3432, 2214, 1648 cm–1, 1H NMR (200 MHz, DMSO–d6) δ 1.21 (t, J = 7.0 Hz, 3H, OCH2CH3), 2.19 (s, 3H, CH3), 3.28 (s, 2H, H–4), 4.18 (q, J = 7.0 Hz, 2H, OCH2CH3), 7.48 (br s, 2H, NH2). 13C (50 MHz DMSO–d6) δ 14.1 (OCH2CH3), 17.8 (CH3), 31.7 (C–4), 58.6 (C–3), 69.7 (OCH2CH3), 97.2 (C–5'), 116.8 (C≡N), 143.6 (C–6'),146.0 (C–2'), 167.1 (C=O). HRMS (EI+) calcd for C10H12N2O3 208.0848, found (M+) 208.0855.
2–Amino–3–cyano–5–ethoxycarbonyl–6–methyl–4–propyl–4H–pyran (4m). Following General Method A, the reaction between 1m (216 mg, 3.0 mmol), malononitrile (2) (198 mg, 3.0 mmol), ethyl acetoacetate (3) (390 mg, 3.0 mmol) and ammonium hydroxide 28% w/w (373 mg, 3.0 mmol) was irradiated with infrared irradiation for 10 min. The crude product was recrystallized from water/ethanol, giving 435 mg, (58%) of 4m as a white solid; mp 163–165 °C; IR (KBr) vmax 3406, 2951, 2186, 1696 cm–1; 1H NMR (500 MHz, DMSO–d6) δ 0.92 (t, J = 7.0 Hz, 3H, H–3'), 1.05–1.45 (m, 7H, H–1', H–2', OCH2CH3), 2.20 (s, 3H, CH3), 3.20 (m, 1H, H–4), 4.20 (m, 2H, OCH2CH3), 6.82 (br s, 2H, NH2). 13C (50 MHz, DMSO–d6) δ 13.7 (C–3'), 13.8 (OCH2CH3), 17.6 (C–3'), 18.0 (CH3), 18.09 (C–2'), 32.2 (C–4), 38.2 (C–1'), 54.5 (C–3), 60.1 (OCH2CH3), 107.8 (C–5), 120.3 (C≡N)), 157.1 (C–6), 160.1 (C–2), 165.8 (C=O). HRMS (EI+) calcd for C13H18N2O3 250.1320, found (M+) 250.1318.
X–ray Structure Study of 4b and 4c. Single crystals were obtained by slow evaporation of concentrated solutions of 4b (EtOH/H2O, white crystals) and 4c (EtOH/H2O, yellow crystals), which were mounted on glass fibers. Crystallographic measurements were performed on an Oxford Diffraction Xcali–bur S single–crystal X–ray liffractometer using Mo KR radiation (graphite crystal monochromator, λ = 71073 A°) at room temperature. Three standard reflections, which were monitored periodically, showed no change during data collection. Unit cell parameters were obtained from least–squares refinement of 26 reflections in the range 2° < 2θ <20°. Intensities were corrected for Lorentz and polarization effects. No absorption correction was applied. Anisotropic temperature factors were introduced for all non–hydrogen atoms. Hydrogen atoms were placed in ilealized positions and their atomic coordinates refined. Structures were solved using the SHELXTL [29], SHELX97 [30], or SIR92 [31] programs as implemented in the WinGX suite [32] and refined using SHELXTL or SHELX97 within WinGX, on a personal computer. In all cases ORTEP and packing diagrams were male with PLATON and ORTEP–3 [33].
Acknowledgements
We thank Bruce Allan Larsen for revising the English of the manuscript. M.A.V. acknowlelges CONCyTEG (Grant 08–16–K662–127 A03) and DAIP (Grant 000156/10) for financial support. F.D. acknowlelges SIP–IPN (Grants 20101131, and 20110175) and CONACYT (Grant 156933) for financial support. J.T. and F.D. are fellows of EDI–IPN and COFAA–IPN programs.
References
1. a) Dömling, A. Chem. Rev. 2006, 106, 17–89. [ Links ] b) Tan, J. N.; Lia, M.; Gu, Y. Green Chem. 2010, 12, 908–914. [ Links ] c) Ugi, I. Pure Appl. Chem. 2001, 73, 187–191 d) Multicomponent Reactions, [ Links ] Zhu, J., Bienaymé, H., Eds.; Wiley–VCH, Weinheim, 2005. [ Links ] e) Cariou, C. C. A.; Clarkson, G. J.; Shipman, M. J. Org. Chem. 2008, 73, 9762–9764. [ Links ]
2. Geen, G. R.; Evans, J. M.; Vong, A. K. In Comprehensive Heterocyclic Chemistry II: Pyrans and their Benzo Derivatives: Applications; Katritzky, A. R.; Rees, C. W.; Scriven, E. F. V. Eds.; Pergamon Press: Oxford, 1996; Vol. 5, 469–500. [ Links ]
3. Jensen, A. A.; Erichsen, M. N.; Nielsen, C. W.; Stensbøl, T. B.; Kehler, J.; Bunch, L. J. Med. Chem. 2009, 52, 912–915. [ Links ]
4. Urbahns, K.; Horváth, E.; Stasch, J. P.; Mauler, F. Bioorg. Med. Chem. Lett. 2003, 13, 2637–2639. [ Links ]
5. Li, A. H.; Ji, X. D.; Kim, H. S.; Melman, N.; Jacobson, K. A. Drug Develop. Res. 1999, 48, 171–177. [ Links ]
6. Kemnitzer, W.; Drewe, J.; Jiang, S.; Zhang, H.; Zhao, J.; Crogan–Grundy, C.; Xu, L.; Lamothe, S.; Gourdeau, H.; Denis, R.; Tseng, B.; Kasibhatla, S.; Cai, S. X. J. Med. Chem. 2007, 50, 2858–2564. [ Links ]
7. Konkoy, C. S; Fick, D. B; Cai, S. X.; Lan, N. C.; Keana, J. F. W. PCT Int. Appl. WO 0075123; Chem. Abstr. 2001, 134, 29313a. [ Links ]
8. a) Martin, N.; Pascual, C.; Seoane, C.; Soto, J. L. Heterocycles 1987, 26, 2811–2816. [ Links ] b) Elnagdi, M. H.; Adbel–Motaleb, R. M.; Mustafa, M.; Zayed, M. F.; Kamel, E. M. J. Heterocycl. Chem. 1987, 24, 1677–1681. [ Links ] c) Wang, X. S.; Zhou, J. X. Zeng, Z. S.; Li, Y. L.; Shi, D. Q.; Tu, S. J. Arkivoc 2006, (xi), 107–113. [ Links ] d) Abdel–Latif, F. F.; Shaker, R. M. Ind. J. Chem. 1990, 29B, 322-325. [ Links ] e) Pratap, U. R.; Jawale, D. V; Netankar, P. D.; Mane, R. A. Tetrahedron Lett. 2011, 52, 5817–5819. [ Links ]
9. Jin, T. S.; Wang, A. Q.; Shi, F.; Han, L. S.; Liu, L. B.; Li, T. S. Arkivoc 2006, (xiv), 78–86. [ Links ]
10. Yadav, J. S.; Sunitha, V.; Subba–Reddy, B. V.; Das, P. P.; Gyan–chander, E. Tetrahedron Lett. 2008, 49, 855–857. [ Links ]
11. Heravi, M. M.; Jani, B. A.; Derikvand, F.; Bamoharram, F.; Oskooie, H. A. Catal. Commun. 2008, 10, 272–275. [ Links ]
12. a) Yi, F.; Peng, Y.; Song, G. Tetrahedron Lett. 2005, 46, 3931–3933. [ Links ] b) Peng, Y.; Song, G. Catal. Commun. 2007, 8, 111–114. [ Links ]
13. Feng, Y. J.; Miao, C. B.; Gao, Y.; Tu, S. J.; Fang, F.; Shi, D–Q. Chin. J. Chem. 2004, 22, 622–626. [ Links ]
14. Peng, Y.; Song, G.; Dou, R. Green Chem. 2006, 8, 573–575. [ Links ]
15. Fotouhi, L.; Fatehi, A.; Heravi, M. M. Int. J. Electrochem. Sci. 2008, 3, 721–726. [ Links ]
16. Anastas, P. T.; Warner, J. C. Green, Chemistry: Theory andPractice; Oxford University Press: Oxford, 1998. [ Links ]
17. Miranda, R.; Noguez, O.; Velasco, B.; Arroyo, G.; Penieres, G.; Martinez, J. O.; Delgado, F. Edu. Quím. 2009, 20, 421–425. [ Links ]
18. a) Delgado, F.; Tamariz, J. Zepeda, G.; Landa, M.; Miranda, R.; García, J. Synth. Commun. 1995, 25, 753–759. [ Links ] b) Obrador, E.; Castro, M.; Tamariz, J.; Zepeda, G.; Miranda, R.; Delgado, F. Synth. Commun. 1998, 28, 4649–4663. [ Links ] c) Alcerraca, G.; Sanabria, R.; Miranda, R.; Arroyo, G.; Delgado, F. Synth. Commun. 2000, 30, 1295–1301. [ Links ]
19. a) Gomez, R. Osnaya, R.; Zamora, I.; Velasco–Bejarano, B., Arroyo, G.; Ramirez–San Juan, E.; Trujillo, J. Delgado F.; Miranda, R. J. Mex. Chem. Soc. 2007, 51, 181–184. [ Links ] b) Osnaya, R.; Arroyo, G.; Parada, L.; Delgado, F.; Trujillo, J.; Salmón, M.; Miranda R. Arkivoc, 2003, (xi), 112–117. [ Links ]
20. Delgado, F.; Cano, A. C.; Garcia, O.; Alvarado, J.; Velasco, L; Alvarez, C.; Rudler, H. Synth Commun. 1992, 22, 2125–2128. [ Links ]
21. a) Penieres, G.; Miranda, R.; García, J.; Aceves, J.; Delgado, F. Heterocycl. Commun. 1996, 2, 401–402. [ Links ] b) Penieres, G.; Garcia, J. G.; Gutierrez, J. L.; Alvarez, C. Green Chem. 2003, 5, 337–339. [ Links ]
22. Vázquez, M. A.; Landa, M.; Reyes, L.; Miranda, R.; Tamariz, J.; Delgado, F. Synth. Commun. 2004, 34, 2705–2718. [ Links ]
23. Martínez, J.; Velasco–Bejarano, B.; Delgado, F.; Pozas, R.; Torres Dominguez, H.; Trujillo, J. Arroyo, G. A.; Miranda, R. Nat. Prod. Commun. 2008, 3, 1465–1468. [ Links ]
24. Pool, G. C.; Teuben, J. H. IR Radiation as a Heat Source in Vacuum Sublimation, in: Practical Organometallic Chemistry, Wayda, A. L.; Darensbourg, M. Y., Eds., Symposium Series 357, Washington, D. C. 1987, pp 30–33. [ Links ]
25. Irradiation was male by an OSRAM R–20 bulb. This bulbe operates at a voltage of 127 V and power of 250 W. The R–20 bulb filament is a wire wound on a spiral type C. The lamp temperature is specified to be 2900 K and their lifetime of 500 h. The bulb of the irradiation source contains a red filter to eliminate the radiation in the far infrared and mil–infrared attenuated considerably. We are conducting a thorough study of the source of this lamp. Preliminary data indicate that the wavelength emitted by the lamp is 1255.6 nm. These data are consistent with information found in the literature,[26] and will be published soon.
26. Shortwave Electric Infrared...The Facts, Copyright© ITW BGK Finishing Systems • 05/03. www.bgk.com. [ Links ]
27. This material is available free of charge via the Internet at the Cambridge Crystallographic Data Centre (e–mail: deposit@ccdc.cam.ac.uk; or http://www.ccdc.cam.ac.uk) as supplementary publication: CCDC 858874 (4b), and CCDC 858875 (4c). [ Links ]
28. a) Babu, N. S.; Pasha, N.; Venkateswara Rao, K. T.; Sai Prasal, P. S.; Lingaiah, N. Tetrahedron Lett., 2008, 49, 2730–2733. [ Links ] b) Kumar, D.; Buchi Reddy, V.; Sharad, S.; Dube, U.; Kapur. S. Eur. J. Med. Chem. 2009, 44, 3805–3809. [ Links ] c) Shestopalov, A. M.; Niaz–imbetova, Z. I.; Evans, D. H.; Niyazymbetov, E. M. Heterocycles 1999, 51, 1101–1107. [ Links ] d) Peng, Y.; Song, G.; Huang, F. Monatsh. Chem. 2005, 136, 727–731. [ Links ]
29. a) SHELXTL, v. 5.10; Bruker AXS, Inc.: Madison, WI, 1998. [ Links ] b) Shedlrick, G. M. Acta Cryst. 2008, A64, 112–122. [ Links ]
30. SHELX97, Programs for Crystal Structure Analysis, Release 97–2; Institüt für Anorganische Chemie der Universität: D–3400 Göttingen, Germany. [ Links ]
31. SIR92. Altomare, A.; Cascarano, G.; Giacovazzo, C.; Guagliardi, A. J. Appl. Cryst. [ Links ]
32. WinGX. Farrugia, L. J. J. Appl. Cryst. 1999, 32, 837–838. [ Links ]
33. a) PLATON. Spek, A. L. J. Appl. Cryst. 2003, 36, 7–13. [ Links ] b) ORTEP–3. Farrugia, L. J. J. Appl. Cryst. 1997, 30, 565. [ Links ]

