










Servicios Personalizados
Revista
Articulo
 Inglés (pdf)
Inglés (pdf)
 Artículo en XML
Artículo en XML Referencias del artículo
Referencias del artículo
 Enviar artículo por email
Enviar artículo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO -
 Accesos
Accesos
Links relacionados
-
 Similares en
SciELO
Similares en
SciELO
Compartir

 Permalink
PermalinkJournal of the Mexican Chemical Society
versión impresa ISSN 1870-249X
J. Mex. Chem. Soc vol.56 no.2 Ciudad de México abr./jun. 2012
Article
Synthesis and Physico–Chemical Characterization of CeO2/ZrO2–SO42– Mixed Oxides
Juan Manuel Hernández–Enríquez,1* Rebeca Silva–Rodrigo,1 Ricardo García–Alamilla,1 Luz Arcelia García–Serrano,2 Brent Edward Handy,3 Guadalupe Cárdenas–Galindo,3 and Arturo Cueto–Hernández4
1 Instituto Tecnológico de Cd. Madero, División de Estudios de Posgrado e Investigación. Juventino Rosas y Jesús Urueta S/N, Col. Los Mangos, 89440 Cd. Madero, Tam., México. 52 833 1160–745. *jmanuelher@hotmail.com
2 Instituto Politécnico Nacional. Centro Interdisciplinario de Investigaciones y Estudios sobre Medio Ambiente y Desarrollo. 30 de Junio #1520, Barrio La Laguna Ticomán, 07340 México D.F., México.
3 CIEP, Facultad de Ciencias Químicas, Universidad Autónoma de San Luis Potosí. Av. Dr. Manuel Nava #6, Zona Universitaria, 78210 San Luis Potosí, San Luis Potosí, México.
4 Universidad Autónoma Metropolitana–Azcapotzalco. Av. San Pablo #180, Col. Reynosa Tamaulipas, Delegación Azcapotzalco, 02200 México D.F., México.
Received September 16, 2011.
Accepted January 28, 2012.
Abstract
Environmentally friendly solid–acid catalysts CeO2/ZrO2–SO42– were prepared by the sol gel method varying CeO2 content (10, 20 and 30 wt%) and using sulfation in situ, maintaining the sulfate ions amount present in the materials at 20 wt%. ZrO2 and ZrO2–SO42–were also prepared for comparison proposes using the same synthesis method. The materials were characterized by X–ray diffraction, nitrogen physisorption, potentiometric titration with n–butylamine, decomposition of 2–propanol and n–pentane isomerization. The specific surface area of ZrO2–SO42– was high (160 m2/g) compared with the unmodified ZrO2 (80 m2/g), however this area decreased with increasing the CeO2 content (37–100 m2/g). There was no significant effect of CeO2 on the tetragonal structure of ZrO2–SO42–. The variation of acid sites amount runs parallel to the change of specific surface area. The acid sites amount decreased with increasing cerium oxide content. The decomposition of 2–propanol results fundamentally in the formation of dehydration products such as propylene and diisopropyl ether, both involving acid sites. In addition, a good performance during the n–pentane isomerization was observed for these materials. The selectivity towards isopentane reaches 84% when the Pt/CeO2/ZrO2–SO42– catalyst with the highest CeO2 content was used.
Key words: Mixed oxides, sol–gel method, sulfation, physico–chemical characterization, acid–base properties.
Resumen
Se prepararon por el método sol–gel catalizadores sólidos–ácidos tipo CeO2/ZrO2–SO42– amigables con el medio ambiente, variando el contenido del CeO2 (10, 20 y 30% peso), usando sulfatación in situ y manteniendo la cantidad de iones sulfato en 20% peso. Utilizando el mismo método y para propósitos de comparación se sintetizaron ZrO2 y ZrO2–SO42–. Los materiales se caracterizaron por difracción de rayos X, fisisorción de nitrógeno, titulación potenciométrica con n–butilamina, descomposición de 2–propanol e isomerización de n–pentano. El área específica de la ZrO2–SO42– fue más alta (160 m2/g) en comparación con la ZrO2 pura (80 m2/g), sin embargo, esta área decrece con el incremento en el contenido de CeO2 (37–100 m2/g). El CeO2 no presento un efecto significativo sobre la estructura tetragonal de la ZrO2–SO42–. La variación en la cantidad de los sitios ácidos disminuye a la par con el cambio de área específica. La cantidad de sitios ácidos decrece con el incremento de óxido de cerio. La descomposición del 2–propanol resulto en la formación de productos de deshidratación, propileno y diisopropil éter, ambos envolviendo sitios ácidos. Los materiales sintetizados presentaron un buen comportamiento durante la isomerización de n–pentano y la selectividad hacia isopentano alcanzó el 84% con el catalizador con mayor contenido de CeO2.
Palabras clave: Óxidos mixtos, método sol–gel, sulfatación, caracterización fisicoquímica, propiedades ácido–base.
Introduction
Mixed oxide catalysts have received tremendous attention both in fundamental and industrial point of view because of their wide spread utility in the chemical industry in comparison with bulk oxide catalysts [1]. The synthesis and development of materials with enhanced thermal, redox and acid–base properties are of paramount interest in designing and selecting catalysts for specific catalytic reactions [2–6]. Recently, a new generation of mixed oxides containing CeO2 and ZrO2 with the features previously mentioned has been developed [7–10]. The ceria–zirconia systems have been recognized as key materials for various applications in catalysis, as ceramics, in fuel cell technology, as solid state electrolytes and as a catalytic promoter in three–way catalysts [11–14]. The selection of an appropriate method of synthesis plays an important role in the success of these materials in the field of catalysis. Several wet–chemical methods, such as hydrothermal synthesis, high–energy mechanical milling, polymerized complex method, coprecipitation, solgel and forced cohydrolysis have been applied to prepare the CeO2–ZrO2 mixed oxides [15–19]. Literature reports indicate that among these synthesis methods, hydrothermal synthesis, polymerized complex method, sol–gel and forced cohydrolysis allow to obtain compositionally homogeneous materials [20]. On the other hand, it can also be noted that besides its redox properties, ceria–zirconia materials present remarkable acid–base properties which have not been investigated extensively. A method to improve the acidic properties of CeO2–ZrO2 materials is the preparation of mixed oxides as CeO2/ZrO2–SO42–. It has been found by means the ammonia–TPD experiments that sulfation of ceria–zirconia mixed oxide can generate the formation of super–acidic sites [21]. Many chemical methods have been developed to measure the acid–base surface properties of the oxides. For this purpose, alkane isomerization and the conversion of secondary alcohols are used [22–25].
The aim of this work is to study the effect of the CeO2 loading on the physico–chemical properties of ZrO2–SO42– and explore the possibility of using CeO2/ZrO2–SO42– as an acid–base bi–functional catalyst.
Results and discussion
Structural properties
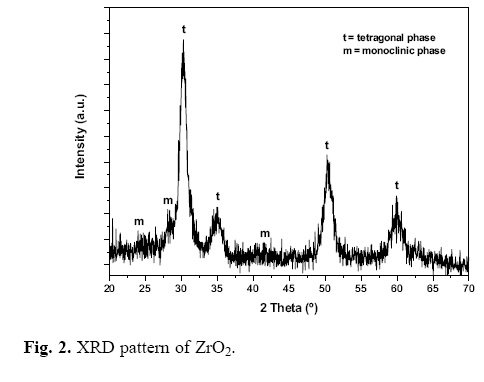
The X–ray diffraction patterns of pure CeO2 and ZrO2 calcined at 600 °C for 3 h are shown in Fig. 1 and Fig. 2, respectively. XRD pattern of CeO2 consists of six main reflections on the 26 scale, 28.57°, 33.00°, 47.60°, 56.39°, 59.13° and 69.51° corresponding to (111), (200), (220), (311), (222) and (400) planes, respectively. The diffraction peaks are characteristic of the cubic ceria phase with fluorite structure (JCPDS card number 34–394). Additional XRD lines assigned to ceria precursor were observed at 2θ = 38.19° and 44.22°. XRD pattern of pure zirconia (Z100) exhibits intense peaks at 26 = 30.26°, 35.10°, 50.42° and 60.04° attributed to the tetragonal phase of zirconia corresponding to (101), (110), (112) and (211) planes (JCPDS card number 80–2155), respectively. In addition, the pattern of zirconia shows others small peaks at 2θ = 24.50°, 28.36° and 41.55°, which are related to the monoclinic phase (JCPDS card number 7–3430). These results suggest that tetragonal and monoclinic phases coexisted in this sample. Fig. 3 displays the diffractograms of the CeO2/ZrO2–SO42– mixed oxides, which clearly exhibit the tetragonal phase as the main one. Introduction of less than 20 wt.% of CeO2 into sulfated zirconia matrix did not involved any appreciable phase modification in the material. However, at CeO2 loadings exceeding 20 wt.%, small peaks at 26 = 25.01°, 26.20° and 31.18° attributed to the monoclinc phase of zirconia were detected. The absence of the characteristic diffracction peaks related to cubic fluorite phase of CeO2 means that the ceria particles were too small to be detected by XRD or the ceria particles were well dispersed on the mixed oxide. The crystallite size of the synthesized materials was determined applying Scherrer's equation, calculated from the full width at half maximum intensity (FWHM) of the mean reflection (101). The average crystallite size of these materials was in the range 6.5–8.5 nm (Table 1).



Textural properties
All synthesized oxides (Z1O2, Z1O2–SO42 and CeO2/ZrO2–SO42–) calcined at 600 °C present the same nitrogen adsorption–desorption isotherms. The nitrogen isotherm and the textural properties of the materials are respectively given in the Fig. 4 and Table 1. The synthesized samples showed a type III isotherm with very reduced hysteresis loop, which is convex whit respect to relative pressure (p/p0) over its entire range [27,28]. Type III isotherms are obtained from non–porous solids or solids with macroporous, though also exist the possibility that the samples have some mesoporous in its morphological structure [29]. The only difference on the isotherms of these materials is in the absorbed volumes, which show different specific surface area results. The first striking difference appears form the comparison between the areas of the pure zirconia (Z100) and sulfated zirconia sample (ZS100). The specific surface area pertaining to ZS100 is about two times larger than the of Z100 calcined at the same temperature. The present larger area of the ZS100 sample can be related to the presence of the SO42– ions which improves the thermal stability into ZrO2 and inhibits the strong syntherization on the material, this result is in agreement with the reported by Yori et al [30]. It has been suggested by Wang et al. that when the sulfuric acid has been used as a synthesis catalyst, the SO42– ions present in the Zr–O framework would replace for OH– ions. Because the thermal stability of the sulfate to the zirconium linkages is much higher than that hydroxyl bridges across two Zr atoms, the removal of SO42– ions needs a higher temperature in comparison with the hydroxyl ions, which delays the material syntherization and also stabilizes the tetragonal phase at low–temperature [19]. The second difference appears from the comparison between the areas of the ZrO2–SO42– and CeO2/ZrO2–SO42– mixed oxides. It can be seen that, for all mixed oxides the specific surface areas were always significantly lower than that of ZrO2–SO42–. The specific surface area decrease as a function of CeO2 content and may be caused due to the formation of agglomerates during the synthesis. On the other hand, the sample with the highest CeO2 content (CeZS70) appeared with an intense yellowish color; it may be inferred to the formation of more CeO2 crystallites on the sample which might reduces the surface mobility of ZrO2–SO24–, thus enhancing the sintering and reducing the surface area.

Acidic properties
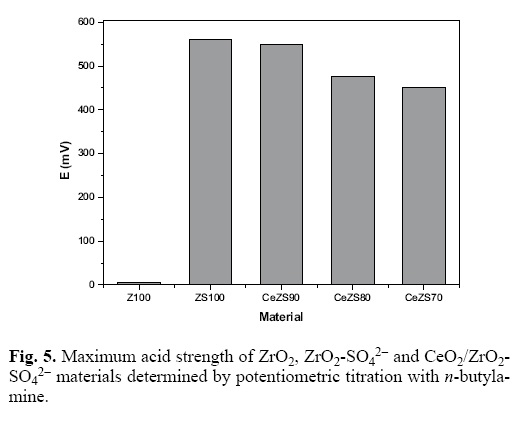
The potentiometric titration method with n–butylamine allowed evaluating the surface acidity of the different synthesized materials. The maximum acid strength (M.A.S.) was determined from the potential reading (mV) at the first point of the titration run. The final amount of titrant used (meq/g–solid) that corresponds to the point at which a plateau is reached indicates the total number of the acid sites [26]. As a criterion for interpreting the results obtained, the acid strength can be assigned according to the following scale: M.A.S. > 100 mV, very strong acid sites; 0 < M.A.S. < 100 mV, strong acid sites; –100 mV < M.A.S. < 0 mV, weak acid sites and M.A.S. < –100 mV, very weak acid sites [31]. In Fig. 5 the remarkable difference in acid strength between pure zirconia (Z100) and sulphated oxides (ZrO2–SO42– and CeO2/ZrO2–SO42–) can be noted from the milivolts range. Pure zirconia is a weakly acidic oxide (E = 5 mV) but it is modified by the SO42– ions addition, which generates strong acidic sites on the zirconia surface according to the scale mentioned by Pizzio et al., [31]. In the case of ZrO2–SO42– (ZS100), the initial potential value was 560 mV; slightly lower than that the corresponding to CeO2/ZrO2–SO42– materials (451–549 mV). From Fig. 6, it can be observed that the addition of Ce2O decrease the number of the acid sites on the ZrO2–SO42– surface. Total acidity decrease in the same order that CeO2 content was increased in the sulfated materials.

2–propanol dehydration
The acid–base properties of ZrO2, ZrO2–SO42– and CeO2–ZrO2–SO42– catalysts are deduced from their catalytic behavior in 2–propanol decomposition and the results are given in Fig. 7. Through the selectivity of this reaction it was deduced which sites predominated in the synthesized materials. As suggest by literature, the dehydration of 2–propanol to produce propylene and diisopropyl ether is assumed to proceed on acidic sites, whereas its dehydrogenation to acetone is presumed to be catalyzed by basic sites in inert atmosphere [32]. It is clear that the variations in catalytic activities of the samples are roughly correlated with the changes of their acid amount sites and with the specific surface area obtained on the catalyst. As can be seen from Fig. 7, the ZrO2–SO42– (ZS100) exhibits a higher catalytic activity than mixed oxides (CeO2/ ZrO2–SO42–). The catalytic activity of all mixed oxides was similar indicating that they contain at the surface a very small concentration of extremely acid sites responsible practically of their lower conversions. The nature, density and strength of surface acid sites of these materials could be related to the CeO2 content. The decomposition of 2–propanol results fundamentally, in the formation of dehydration products such as propylene and diisopropyl ether, both involving acid sites. Depending on the CeO2 content in the catalytic supports, the selectivity to diisopropyl ether fluctuated in the range between 76 and 96%. Pure zirconia (Z100) was not able to transforms the alcohol at the same temperature than the sulfated materials. However, Z100 catalyzed the dehydration of 2–propanol to propylene at 120 °C with very low conversion (0.11%). In accordance with the selectivity pattern observed in Fig. 7, it can be concluded that all synthesized materials had an acid character. Even though it has been reported that ZrO2 presents amphoteric properties [33], in this work there was not acetone production which indicated no basic sites in the materials or if they existed, they do not have the sufficient strength to direct the reaction toward the dehydrogenation of the alcohol.

n–pentane isomerization
The catalytic properties of Pt/ZrO2–SO42– and Pt/CeO2/ZrO2–SO42– mixed oxide catalysts were tested in the n–pentane isom–erization reaction at 250 °C and atmospheric pressure. Fig. 8 shows conversion and products selectivity in this reaction for these materials. Pt/ZrO2 is not active for the reaction; however, Pt/ZrO2–SO42– and all Pt/CeO2/ZrO2–SO42– mixed oxide catalysts show catalytic activities with a product distribution where iso–pentane is a main product and n–propane, n–butane, iso–butane also were observed in smaller amounts. These results show that this reaction is a typical acid–catalyzed reaction and these solid acid catalysts are effective for this reaction. The activity order of all Pt/CeO2/ZrO2–SO42– catalysts is as follows: Pt/ZS100 > Pt/CeZS90 > Pt/CeZS80 > Pt/CeZS70. For Pt/CeO2/ ZrO2–SO42– catalysts, the catalytic activity reduce gradually with increased CeO2 loading, in agreement with the decrease in the specific surface area and surface acidity. The high activity of the Pt/ZrO2–SO42– (Pt/ZS100) catalyst may be ascribed to its excellent total acidity and tetragonal crystalline ZrO2 phase. Although it has been reported by many researches that the sul–fated ZrO2 of metastable tetragonal phase has higher catalytic activity than the monoclinic phase in many reactions such as the isomerization of n–paraffins [34–39], it is more important to consider the relation between the presence of crystallite defects and the stabilization of the tetragonal structure. Tetragonal structure in catalyst promoted in the state of amorphous gel is a consequence of the stabilization of anionic vacancies by sulfate ions. Catalytic activity is supposedly related to charge abstraction and stabilization from radical and ionic intermediates, a process enabled or enhanced by anionic vacancies in tetragonal zirconia catalysts [40]. On the other hand, for all catalysts a steady state is reached after a transitory period of 60 min, characterized by an increase of the catalytic activity (Fig. 9). At the same time, the selectivity of isomerization product grows. The induction period observed might be attributed to the formation of the reaction intermediates on the catalyst surface. After of the transitory time, the conversion was constant with the increase of time, which indicates a good catalytic stability in the solids at the evaluated conditions reactions, may be due to Pt presence. Tomishige et al. have suggested that molecular hydrogen fed to the isomerization reaction dissociates on the platinum to hydrogen atoms, which undergo spillover onto the acid support and convert to an H+ and an e– or H–. The role of spilt–over hydrogen is the regeneration of Brönsted acid sites and the supply of hydride to carbenium ion intermediate before ß–scission events occur, avoiding cracking and polymerization reactions and thus Pt induces the reaction stability [41].


In Fig. 8 it can be observed that the selectivity to iso–pen–tane was higher than 70% for all tested materials. The maximum selectivity toward the main product (i–C5) was obtained when the CeO2 content reached 30 wt.% in the catalyst, however, this material present the lowest conversion. The promoter effect of CeO2 on the selectivity of this reaction is probably due to the decrease of density and strength of the surface acid sites. The cause of the promotion effect is still not clear.
Conclusions
The results showed that the interaction between sulphated zir–conia and ceria influenced the physicochemical properties of synthesized mixed oxides. The presence of ceria and sulfate ions delays the phase transition of ZrO2 from tetragonal to monoclinic. The specific surface area and acidity of mixed oxides decrease in proportion to the ceria content. There was a good correlation between catalytic activity and acidity for the probe reactions, 2–propanol dehydration and n–pentane isom–erization. The probe reactions provide strong evidence that the surface of the mixed oxide had an acid character. Specifically 2–propanol conversion reaction, results fundamentally in the formation of dehydration products such as propylene and di–isopropyl ether, both involving acid sites. On the other hand, in n–pentane isomerization, the cracking products were greatly inhibited, while a large amount of iso–pentane was produced on the moderate acidic surface of mixed oxides. The best conversion of n–pentane could be as high as 16% over the Pt/CeZS90 catalyst and the highest selectivity towards iso–pentane formation (84%) was obtained with the material Pt/CeZS70.
Experimental
Mixed oxides preparation
The mixed oxides CeO2/ZrO2–SO42– investigated in this work were synthesized by the sol–gel method, varying the CeO2 content (10, 20 and 30 wt.%). The sol–gel synthesis was carried out as follows: Cerium acetate [(CH3CO2)3Ce • H2O; Sig–ma–Aldrich; 99.99%] was previously dissolved in a mixture of tert–butyl alcohol [(CH3)3COH; Sigma–Aldrich; 99.9%)] and deionized water. After that, the mixture was vigorously stirred by means of a magnetic stirrer and refluxed at 70 °C for 1 h. A predetermined amount of zirconium n–butoxide (Zr[O(CH2)3CH3]4; Sigma–Aldrich; 80 wt.% in 1–butanol) dissolved in tert–butyl alcohol was added drop–wise and the system was maintained in continuous stirring and reflux at 70 °C for 2 h. During the last step, sulfation was carried out in situ using concentrated H2SO4 as sulfating agent, maintaining the sulfate ions amount constant at 20 wt.% on the final material. The resulting gel was then aged for 24 h and dried in static conditions at 120 °C for 24 h. The synthesized materials in powder form were calcined in a dynamic air atmosphere at 600 °C for 3 h. Pure and sulfated zirconia (ZrO2 and ZrO2–SO42–) were also prepared for comparison proposes using the same synthesis method. Synthesized mixed oxides were named according to the sulfation agent and the weight percentage of CeO2 present in the material, its nomenclature is shown in the Table 1.
For the n–pentane isomerization, the mixed oxides CeO2/ ZrO2–SO42– were impregnated with platinum (Pt) by means of incipient wetness method from a platinum acetylacetonate solution [Pt(C5H7O2)2; Sigma–Aldrich; 99.99%] followed by drying at 120 °C for 6 h and calcination in air flow at 400 °C for 3 h. The theoretical concentration of Pt was fixed in 0.3 wt.% for all catalysts.
Physico–chemical characterization
Powder X–ray diffraction patterns were recorded on a Bruker diffractometer using Cu Ka radiation (X = 1.5406 Â) and a graphite secondary beam monochromator; the intensities of the diffracction lines were obtained in the 26 range between 20 and 70° with a step size of 0.02° and a measuring time of 2.7 s per point. The crystallite size of the materials was determined by the Scherrer's equation.
Nitrogen physisorption was used to determine the specific surface areas of the materials at the temperature of liquid nitrogen (–196 °C) in a Quantachrome Autosorb–1 instrument. Prior to the measurements, samples were outgassed at 350 °C for 2 h. The specific surface area was calculated using the BET equation and the BJH method was used to calculate the average pore diameter and the pore volume of the samples.
Materials acidity was determined by potentiometric titration with n–butylamine. A small quantity of 0.025 M n–butyl–amine in acetonitrile was added to a known mass of solid, and agitated for 3 h. Later, the suspension was titrated with the same base at 0.2 ml/min. The electrode potential variation was measured with a JENWAY–3310 digital pH meter. The voltage is recorded at intervals as the titrant is added and plotted against the amount of base used. The potentiometric titration method reported in this study has been described in detail previously by Cid and Pecchi [26].
2–propanol dehydration was carried out in a fixed–bed reactor operating at 80 °C and atmospheric pressure. Approximately 0.1 g of the catalyst was loaded into the reactor. Prior to the reaction, the catalyst was preheated at 350 °C for 1 h in a purified nitrogen flow. During the reaction, an effluent collected periodically was analyzed by Varian 3400 gas chromatograph equipped with a FID detector and a column packed with Carbo–wax 1540 on Chromosorb (1.5 m length and 3.18 mm I.D.).
The n–pentane isomerization was carried out in a fixed–bed reactor, operating at 250 °C and atmospheric pressure. The catalyst (0.3 g) was reduced at 350 °C for 1 h in a flow of hydrogen prior to running the test reaction. The products were analyzed on–line by a Varian 3400 gas chromatograph equipped with a FID detector and a column packed with 23 SP–1700 on 80/100 Chromosorb (6 m length and 3.18 mm I.D.).
References
1. Burri, D. R.; Choi, K. M.; Lee, J. H.; Han, D. S.; Park, S. E. Catal. Commun. 2007, 8, 43–48. [ Links ]
2. Brayner, R.; Ciuparu, D.; da Cruz, G. M.; Fiévet, F.; Bozon, F. Catal. Today 2000, 57, 261–266. [ Links ]
3. Reddy, B. M.; Sreekanth, P. M.; Lakshmanan, P.; Khan, A. J. Mol. Catal. A: Chem. 2006, 244, 1–7. [ Links ]
4. Liu, Ch.; Luo, L.; Lu, X. Kinet. Catal. 2008, 49, 676–681. [ Links ]
5. Ji, Y.; Toops, T. J.; Crocker, M. Catal. Lett. 2009, 127, 55–62. [ Links ]
6. Gao, X.; Jiang, Y.; Fu, Y.; Zhong, Y.; Luo, Z.; Cen, K. Catal. Commun. 2010, 11, 465–469. [ Links ]
7. Takeguchi, T.; Furukawa, S.; Inoue, M. J. Catal. 2001, 202, 1424. [ Links ]
8. Bedrane, S.; Descorme, C.; Duprez, D. Catal. Today 2002, 75, 401–405. [ Links ]
9. Zhao, M.; Shen, M.; Wang, J. J. Catal. 2007, 248, 258–267. [ Links ]
10. Mejri, I.; Younes, M. K.; Ghorbel, A.; Eloy, P.; Gaigneaux, E. M. J. Porous Mater. 2009, 16, 745–748. [ Links ]
11. Bozo, Ch.; Guilhaume, N.; Herrmann, J. M. J. Catal. 2001, 203, 393–406. [ Links ]
12. Prasad, D. H.; Lee, J. H.; Lee, H. W.; Kim, B. K.; Park, J. S. J. Ceramic. Process. Res. 2009, 10, 748–752. [ Links ]
13. Haneda, M.; Houshito, O.; Sato, T.; Takagi, H.; Shinoda, K.; Nakahara, Y.; Hiroe, K.; Hamada, H. Catal. Commun. 2010, 11, 317–321. [ Links ]
14. Vagia, E. Ch.; Lemonidou, A. A. J. Catal. 2010, 269, 388–396. [ Links ]
15. Thammachart, M.; Meeyoo, V.; Risksomboon, T.; Osuwan, S. Catal. Today 2001, 68, 53–61. [ Links ]
16. Suda, A.; Ukyo, Y.; Sobukawa, H.; Sugiura, M. Rev. Toyota CRDL 2002, 3, 6–13. [ Links ]
17. Adamski, A.; Djéga, G.; Sojka, Z. Catal. Today 2007, 119, 120–124. [ Links ]
18. Reddy, B. M.; Patil, M. K.; Lakshmanan, P. J. Mol. Catal. A: Chem. 2006, 256, 290–294. [ Links ]
19. Wang, J. A.; González, G.; Chen, L.; Valenzuela, M. A.; Moran, M.; Vázquez, A.; Castillo, S. React. Kinet. Catal. Lett. 2007, 90, 381–387. [ Links ]
20. Lascalea, G. E.; Llamas, D. G.; Djurado, E.; Walsöe de Reca, N. E. J. Arg. Chem. Soc. 2003, 91, 135–142. [ Links ]
21. Reddy, B. M.; Sreekanth, P. M.; Lakshmanan, P.; Khan, A. J. Mol. Catal. A: Chem. 2006, 244, 1–7. [ Links ]
22. Ouqour, A.; Coudurier, G.; Vedrine, J. C. J. Chem. Soc. Faraday Trans. 1993, 89, 3151–3155. [ Links ]
23. Tomczak, D. C.; Allen, J. L.; Poeppelmeier, K. R. J. Catal. 1994, 146, 155–165. [ Links ]
24. Rekoske, J. E.; Barteau, M. A. J. Catal. 1997, 165, 57–72. [ Links ]
25. Moles, P. in: Zirconium Compounds in Catalysis: Current and Future Uses, Technical Report MEL CHEMICALS 1999, http://www.zrchem.com. [ Links ]
26. Cid, R.; Pecchi, G. Appl. Catal. 1985, 14, 15–21. [ Links ]
27. Roskar, R.; Kmetec, V. Chem. Pharm. Bull. 2005, 53, 662–665. [ Links ]
28. Klewicki, R.; Konopacka, D.; Uczciwek, M.; Irzyniec, Z.; Pias–ecka, E.; Bonazzi, C. J. Hortic. Sci. Biotech. 2009, 84, 75–79. [ Links ]
29. Kalliopi A. Pore structure of cement–based materials. Testing, interpretation and requirements. Ed. Taylor and Francis Group, 2006. [ Links ]
30. Yori, J. C.; Pieck, C. L.; Parera, J. M. Catal. Lett. 2000, 64, 141–146. [ Links ]
31. Pizzio, L.; Vázquez, P.; Cáceres, C.; Blanco, M. Catal. Lett. 2001, 77, 233–239. [ Links ]
32. Petre, A. L.; Auroux, A.; Gervasini, A.; Caldarau, M.; Ionescu, N. I. J. Therm. Anal. 2001, 64, 253–260. [ Links ]
33. Chary, K. V. R.; Seela, K. K.; Naresh, D.; Ramakanth, P. Catal. Commun. 2008, 9, 75–81. [ Links ]
34. Jin, T.; Yamaguchi, T.; Tanabe, K. J. Phys. Chem. 1986, 90, 4797–4805. [ Links ]
35. Morterra, C.; Cerrato, G.; Pina, F.; Signorreto, M. J. Catal. 1995, 157, 109–123. [ Links ]
36. White, R. L.; Sikabwe, E. C.; Coelho, M. A.; Resasco, P. E., J. Catal. 1995, 157, 755–758. [ Links ]
37. Yadav, G. D.; Nair, J. J. Micropor. Mesopor. Mater. 1999, 33, 1–48. [ Links ]
38. Yori, J. C.; Parera J. M. Catal. Lett. 2000, 65, 205–208. [ Links ]
39. Stichert, W.; Schüth, F.; Kuba, S.; Knözinger, H. J. Catal. 2001, 198, 277–285. [ Links ]
40. Vera, C. R.; Pieck, C. L.; Shimizu, K.; Parera J. M. Appl. Catal. A. 2002, 230, 137–151. [ Links ]
41. Tomishige, K.; Akihiro, O.; Fujimoto, K. Appl. Catal. A. 2000, 194–195, 383–393. [ Links ]

