










Servicios Personalizados
Revista
Articulo
 Inglés (pdf)
Inglés (pdf)
 Artículo en XML
Artículo en XML Referencias del artículo
Referencias del artículo
 Enviar artículo por email
Enviar artículo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO -
 Accesos
Accesos
Links relacionados
-
 Similares en
SciELO
Similares en
SciELO
Compartir

 Permalink
PermalinkJournal of the Mexican Chemical Society
versión impresa ISSN 1870-249X
J. Mex. Chem. Soc vol.56 no.2 Ciudad de México abr./jun. 2012
Article
Density Functional Theory Analysis of Borazyne Complexes of Ni(B3N3HnF2–n) (CO)2 (n = 0–2)
Reza Ghiasi and Amir Hossein Hakimyoon
Department of Chemistry, Basic Science Faculty, East Tehran Branch, Qiam Dasht, Tehran, Islamic Azad University, Tehran, Iran. rezaghiasi1353@yahoo.com, rghyasi@qdiau.ac.ir
Received August 9, 2011.
Accepted November 19, 2011.
Abstract
The electronic structure and properties of Ni(B3N3HnF2–n) (CO)2 (n = 0–2) complexes have been explored using hybrid density functional B3LYP theory. Calculations indicate B–fluorinated isomers are more stable, less polarizable, and harder than N–fluorinated isomers. The aromatic nature of the borazyne rings have been analyzed by nucleus independent chemical shift (NICS). The atoms in molecules (AIM) analysis indicates that Ni–Ccarbonyl bonds distance is well correlated with the electron density of critical point (ρrcp) in all species.
Key words: Borazyne, borazyne complexes, aromaticity, nucleus–independent chemical shift (NICS), quantum theory atoms in molecules methodology (QTAIM).
Resumen
La estructura electrónica y propiedades de los complejos Ni (B3N3HnF2–n)(CO2)(n = 0–2) han sido explorados usando la teoría de funcionales de la densidad de híbridos B3LYP. Los cálculos indican que los isómeros B–fluorados son más estables, menos polarizables, y más duros que los isómeros N–fluorados. La naturaleza aromática de los anillos de borazina ha sido analizada por desplazamiento químico independiente del núcleo. El análisis de átomos en moléculas indica que las distancias de enlace Ni–Ccarbonilo correlaciona bien con la densidad electrónica del punto crítico (ρprep) en todas las especies.
Palabras clave: Borazina, complejos de borazina, aromaticidad, desplazamiento químico independiente del núcleo, átomos y moléculas.
Introduction
The structure and properties of benzyne have been studied theoretically and experimentally for many years [1–4]. The replacement of CC by BN is known to lead to Borazyne that suggested as an intermediate in the formation of borazanaphthalene and diborazine, during the photolysis of borazine [5]. Borazyne has been not isolated and characterized. A few investigations have been reported about it [6]. Because of the cinsiderable difference between the electronegativity of boron and nitrogen, the ring delocalization of electrons in the borazyne ring is weakened.
Since the first attempt for making metal complexes using benzyne by Wittig and Bickelhaupt in 1958 [7], many benzyne complexes have been successfully prepared [8–11]; for example, M. A. Bennett et al. [12] synthesized organometallic compounds, NiL2(C6H4)(L = PCy3, PiPr3, Cy = cyclohexyl, iPr = isopropyl). Deaton and Gin studied the reactions of nickel(0)–benzyne complexes with symmetrically substituted 1,3–diynes in the presence of triethylphosphine, which lead to the regiose–lective formation of 2,3–dialkynyl naphthalenes [13].
In the present study, the quantum chemical methods were used in order to gain a deeper insight into the structure and bonding of Ni(B3N3HnF2–n) (CO)2 (n = 0–2) complexes and phenomena of the substituent effect in a benzyne ring.
Computational Methods
All calculations were carried out with the Gaussian 03 suite of program [14]. All molecules were described by the standard 6–31G(d,p) basis set [15–17]. Geometry optimization was performed using Beckes hybrid three–parameter exchange functional and the nonlocal correlation functional of Lee, Yang, and Parr (B3LYP) [18]. A vibrational analysis was performed at each stationary point which corresponds to an energy minimum.
The nucleus–independent chemical shift (NICS) [19–20] has been defined as the absolute magnetic shielding computedat the center of a ring in a molecule. NICS(0.0), NICS(0.5), NICS(1.0), NICS(1.5) and NICS(2.0) were calculated at 0 (center), 0.5, 1.0, 1.5, and 2.0 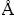 above the ring, respectively.
above the ring, respectively.
The AIM2000 program [21] was used fir the topological analysis of electron density, and the characteristics of ring critical points (RCPs) were taken into account: density at RCP (p(rc)), and its Laplacian ( 2p(rc)).
2p(rc)).
The two–center delocalization index, δ(A, B), was originally defined by Bader as a measure of the extent of the correlative interaction between electrons into different regions [22]. The term δ(A, B), gives a quantitative idea of the number of electrons delocalized or shared between atoms A and B. For a closed shell system the delocalization index can be expressed as

where SAij is the overlap between doubly occupied orbitals i and j iver the basin of atom A. Using the delocalization index, the average two center indices (ATI) is defined as [23]

where the summation runs over all adjacent pairs of atoms around 6–membered ring.
Results and discussion
Isomers stability
Relative energies (ΔE), pilarizability (α), and HOMO–LUMO gaps energies of all species are summarized in Table 1. Calculations indicate the B–fluorinated isomers are more stable, less polarizable, and harder than N–fluorinated isomers. As expected frim the principles of minimum energy, minimum polarizability, and maximum HOMO–LUMO gaps, that is, when an isomer changes from the most stable to other less stable species in most cases, the energy increases, the HOMO–LUMO gaps decreases, and the polarizability increases [24]. The increased stability of the B–fluorinated molecules is due to lone pair/lone pair electron repulsion in the N–F bond.

Thermochemical Analysis
Thermichemical analysis is dine for borazyne complexes with the following reaction:
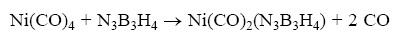
The values of ΔH, ΔS and ΔG are reported in Table 2 in which the individual terms are referred to a temperature of 298 K. As can be verified, the ΔS values are quite similar for all complexes. Also, it is obvious that ΔS should be positive, since in this reaction three particles formed. Although the relative difference of the ΔG are almist the same as the ΔH. The equilibrium constants of the all complexes are given in Table 2. This shows that the equilibrium constant is more for the fluorinated complexes. This trend is compatible with the decreasing of Ni–b and Ni–N bonding length in fluorinated complexes (Figure 1).


Geometries
The main parameters of geometric structures optimized are indicated in Figure 1. The B1–N1 bond distances of borazyne and fluorinated borazynes becimes elongated when benzyne forms a complex with Ni(CO)2.
The Ni–CO (trans to B) bond lengths in all complexes are slightly linger than the Ni–CO (trans ti N). The N atom is more weakly bonded than B (Figure 1), and thus, they induce stringer Ni–COtrans binds in complexes. The M–CO σ–bond is formed by the dination of a lone pair of carbon atom into the d orbital of the metal. Overlapping if filled d orbital of metal with the empty CO π* orbital results in the back bonding. The geometrical parameters show the increasing of Ni–CO bond distances in fluorinated borazynes. This increasing is compatible with back bonding decreasing in Ni–CO bond.
Nucleus Independent Chemical Shift analysis (NICS)
NICS is an easy and efficient criterion to identify aromatic nature. A large negative NICS at the ring center (or inside and above the molecular plane) implies the presence of diamagnetic ring currents.
As shown in Table 3, all the negative computed NICS(0.0) values at the geometrical centers of ring suggests that these rings are obviously aromatic. In order to further identify the aromaticity, we calculated the NICS values (including NICS(0.5), NICS(1.0), NICS(1.5), and NICS(2.0)) by placing a series of ghost atoms above (by 0.5, 1.0, 1.5, 2.0 ) the geometrical centers. All these NICS values are mainly attribute to the delocalized π electrons current. They are shown in Table 3 in detail and are all negative. Also, all NICS(0.0), NICS(0.5), and NICS(1.0) values are most negative of di, moni, and non–fluorinated species, respectively. The negative values inside and above the rings adequately prive that the diamagnetic ring current effect, characteristic for aromaticity, exists in these ground states. It is likely that induced magnetic fields generated by the σ aromaticity are particularly large in the center of the ring, whereas systems having π arimaticity show a minimum NICS at certain distances from the center of the ring, like in benzene.
On the other hand, the NICS values present the increasing of aromaticity in fluorinated rings. Furthermore, aromaticity of N–fluorinated rings is more than B– fluorinated ones. The comparison of NICS values of free borazyne and borazyne complexes show that coordination of Ni(CO)2 decreases aromaticity of ring. The HOMO–LUMO gap values present the similar result. Therefire, magnetic criteria of aromaticity is compatible with the electronic aromaticity criteria.
QTAIM analysis
As it is difficult to separate the σ and π contributions to the electron density at the bond critical point, the p(r) values can be used to evaluate bond strength for different types of bonds (Table 4). The comparison of electron density in the bond critical points of B1N1 shows that p(B1N1) decreases in fluorinated rings. This trend is well–matched with the results of the geometrical analysis.

The different values of p(r) and 2p(r) for the Ni–Ccarbonyl bonds evidently indicate the relative Ni–C bond strengths. This result is in agreement with the geometrical analysis, showing that the Ni–C bonds of H4 are shorter than other species.
The value of electron density and its Laplacian estimated at bond critical point of Ni–Ccarbonyl correlate very well with the strength of the bond, as well as with its length, since, as it is well known, both the strength and length of a bond are mutually dependent. A good relationship is present between p(Ni–Ccarbonyl) values and r(NiCcarbonyl)(R2 = 0.990 for Ni–C trans ti B, and R2 = 0.999, for Ni–C trans ti N).
Interestingly, in the case of all the Ni–Ccarbonyl bonds, 2p values at cirrespinding BCPs are positive, as it was round for closed–shell interactions, but with H(p) < 0, as found for shared interactions. This is in agreement with observations made for the Ti–C bonds in titanium complexes [25] and similar complexes [26], in the case when the metal–ligand bonding has a characteristic that represents a mix of the closed–shell and shared parameters.
Additionally, the H(p) values are more negative for Ni–Ccarbonyl (trans ti N) bonds, which is directly connected with relative greater predominance of lV(p)l magnitude over the G(p) magnitude. This suggests a more covalent character of the Ni–Ccarbonyl (trans to N) bonds as compared with the trans to B ones.
Generally, the greater value of lH(q)l (with negative sign), the more covalent character of the bond. It seems therefore that the covalent character of the Ni–CO bonds increases in bonds that are trans to N.
Table 5 shows the delicalization indexes (DI) and the average two center indices (ATI) for all species. It has to be noticed, the ATI values in N–fluorinated rings are more than B–fluorinated ones. The similar trend has been found in NICS values.
Conclusion
In this paper were investigated the structures and bonding of the Ni(B3N3HnF2–nNi(CO)2 (n = 0–2) complexes. The energetic results suggest that B–fluorinated isomers are most stable among the mono–, di–fluorinated complexes. The NICS calculations confirmed the aromaticity in the borazyne rings of the compounds. Using the analyses of both electron densities and energy densities, we ciuld explain the characters of the Ni–C bonds in complexes.
References
1. Proft, F. D.; Schleyer, P. v. R.; Lenthe, J. H. v.; Stahl, F.; Geerlings, P. Chem. Eur. J. 2002, 8, 3402–3410. [ Links ]
2. Kraka, E.; Anglada, J.; Hjerpe, A.; Filatov, M.; Cremer, D. Chem. Phys. Lett. 2001, 348, 115–125. [ Links ]
3. Nelson, E. D.; Artau, A.; Price, J. M.; Tichy, S. E.; Jing, L.; Kenttamaa, H. J. Phys. Chem. A 2001, 705, 10155 –10168. [ Links ]
4. Crawford, T. D.; Kraka, E.; Stanton, J. F.; Cremer, D. J. Chem. Phys. 2001, 114, 10638 –10650. [ Links ]
5. Neiss, M. A.; Porter, R. F. J. Am. Chem. Soc. 1972, 94, 1438–1443. [ Links ]
6. Fazen, P. J.; Burke, L. A. Inorg. Chem. 2006, 45, 2494–2500. [ Links ]
7. Wittig, G.; Bickelhaupt, F.: Chem. Ber. 1958, 91, 883. [ Links ]
8. Andino, J. G.; Kilgore, U. J.; Pink, M.; Ozarowski, A.; Krzystek, J.; Telser, J.; Baik, M.–H.; Mindiola, D. J.: Chem. Sci. 2010, 1, 351–356. [ Links ]
9. Bennett, M. A.; Schwemlein, H. P. Angew. Chem. Int. Ed. Engl. 1989, 28, 1296– 1320. [ Links ]
10. Hughes, R. P.; Laritchev, R. B.; Williamson, A.; Incarvito, C. D.; Zakharov, L. N.; Rheingold, A. L. Organometallics 2002, 21, 4873–4885. [ Links ]
11. Retbøll, M.; Edwards, A. J.; Rae, A. D.; Willis, A. C.; Bennett, M. A.; Wenger, E. J. Am. Chem. Soc. 2002, 124, 8348–8360. [ Links ]
12. Bennett, M. A.; Hamhley, T. W.; Robertson, N. K. Organometallic 1985, 4, 1992. [ Links ]
13. Deaton, K. R.; Gin, M. S. Organometallic 2003, 5, 2477–2480. [ Links ]
14. Frisch, M. J.; Trucks, G. W.; Schlegel, H. B.; Scuseria, G. E.; Robb, M. A.; Cheeseman, J. R.; Montgomery, J. A.; Jr.; Vreven, T.; Kudin, K. N.; Burant, J. C.; Millam, J. M.; Iyengar, S. S.; Tomasi, J.; Barone, V.; Mennucci, B.; Cossi, M.; Scalmani, G.; Rega, N.; Petersson, G. A.; Nakatsuji, H.; Hada, M.; Ehara, M.; Toyota, K.; Fukuda, R.; Hasegawa, J.; Ishida, M.; Nakajima, T.; Honda, Y.; Kitao, O.; Nakai, H.; Klene, M.; Li, X.; Knox, J. E.; Hratchian, H. P.; Cross, J. B.; Adamo, C.; Jaramillo, J.; Gomperts, R.; Stratmann, R. E.; Yazyev, O.; Austin, A. J.; Cammi, R.; Pomelli, C.; Ochterski, J. W.; Ayala, P. Y.; Morokuma, K.; Voth, G. A.; Salvador, P.; Dannenberg, J. J.; Zakrzewski, V. G.; Dapprich, S.; Daniels, A. D.; Strain, M. C.; Farkas, O.; Malick, D. K.; Rabuck, A. D.; Raghavachari, K.; Foresman, J. B.; Ortiz, J. V.; Cui, Q.; Baboul, A. G.; Clifford, S.; Cioslowski, J.; Stefanov, B. B.; Liu, G.; Liashenko, A.; Piskorz, P.; Komaromi, I.; Martin, R. L.; Fox, D. J.; Keith, T.; Al–Laham, M. A.; Peng, C. Y.; Nanayakkara, A.; Challacombe, M.; Gill, P. M. W.; Johnson, B.; Chen, W.; Wong, M. W.; Gonzalez, C.; Pople, J. A.: Gaussian 03. Revision B.03 ed.; Gaussian, Inc., Pittsburgh PA, 2003. [ Links ]
15. Ditchfield, R.; Hehre, W. J.; Pople, J. A. J. Chem. Phys. 1971, 54, 724. [ Links ]
16. Hehre, W. J.; Ditchfield, R.; Pople, J. A. J. Chem. Phys. 1972, 56, 2257. [ Links ]
17. Rassolov, V. A.; Ratner, M. A.; Pople, J. A.; Redfern, P. C.; Curtiss, L. A. J. Comp. Chem. 2001, 22, 976. [ Links ]
18. Becke, A. D. J. Chem. Phys. 1993, 98, 5648–5652. [ Links ]
19. Chen, Z.; Wannere, C. S.; Cirminboeuf, C.; Puchta, R.; Schleyer, P. v. R. Chem. Rev. 2005, 105, 3842–3888. [ Links ]
20. Schleyer, P. v. R.; Maerker, C.; Dransfeld, A.; H.Jiao; Himmes, N. J. R. v. E. J. Am. Chem. Soc. 1996, 118, 6317–6318. [ Links ]
21. Bader, R. F. W.: AIM2000 Program. ver 2.0, ed.: Hamilton, Mc–Master University, 2000. [ Links ]
22. Bader, R. F. W.; Stephens, M. E. J. Am. Chem. Soc. 1975, 97, 7391–7399. [ Links ]
23. Fradera, X.; Austen, M. A.; Bader, R. F. W. J. Phys. Chem. A 1999, 103, 304–314. [ Links ]
24. Roy, D. R.; Chattaraj, P. K. J. Phys. Chem. A 2008, 112, 1612–1621. [ Links ]
25. Bader, R. F. W.; Matta, C. F. Inorg. Chem 2001, 40, 5603. [ Links ]
26. Palusiak, M. J. Organomet. Chem 2007, 692, 3866–3873. [ Links ]

