










Servicios Personalizados
Revista
Articulo
 Inglés (pdf)
Inglés (pdf)
 Artículo en XML
Artículo en XML Referencias del artículo
Referencias del artículo
 Enviar artículo por email
Enviar artículo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO -
 Accesos
Accesos
Links relacionados
-
 Similares en
SciELO
Similares en
SciELO
Compartir

 Permalink
PermalinkTropical and subtropical agroecosystems
versión On-line ISSN 1870-0462
Trop. subtrop. agroecosyt vol.14 no.2 Mérida may./ago. 2011
Artículos de investigación
Molecular pattern of Mycobacterium bovis isolates and its relationship with risk factors associated with the presence of bovine tuberculosis in northern Mexico
Patrón molecular de aislamientos de Mycobacterium bovis y su asociación con algunos factores de riesgo asociados con la presencia de tuberculosis bovina en el noreste de México
Isaac F. Padilla-Campos1, José C. Segura-Correa1, Jorge C. Rodríguez-Buenfil1, Alberto Morales-Loredo2, Genoveva Alvarez-Ojeda3
1 Campus de Ciencias Biológicas y Agropecuarias, Universidad Autónoma de Yucatán. Km 15.5 carretera Mérida-Xmatkuil, Apartado postal 4-116, Mérida, Yucatán, México. *Corresponding Author E-mail: scorrea@uady.mx.
2 Consorcio Técnico del Noreste de México-Universidad Autónoma de Nuevo León, Km 4.5 Carretera a Reynosa-Guadalupe, CP 67100 Nuevo León, México.
3 Instituto Nacional de Investigaciones Forestales, Agrícolas y Pecuarias- Centro de Investigación Regional del Noreste, carretera Matamoros Reynosa km 61 CP 88900, Río Bravo Tamaulipas, México.
Submitted May 26, 2010
Accepted November 29, 2010
Revised received January 27, 2011
Abstract
The objective of this study was to determine the molecular pattern of M. bovis isolates from cattle of Northern Mexico and its relationship with some risk factors. Isolates (n=60) were obtained from the states of Coahuila (COA, n=14), Tamaulipas (TAM, n=16), Nuevo Leon (NL, n=14) and, Baja California and Durango (DUR, n=16). The risk factors studied were: system of production (Dairy and beef), state, age, lesion type (localized and generalized), and type of presentation (caseous and calcified). Samples were analyzed at the Regional Laboratory of Monterrey NL, following a spoligotyping protocol. Twenty-five spoligotypes belonging to the M. bovis complex were identified. Eleven (18.3%) isolates presented a unique pattern, whereas 49 (81.7%) were grouped in 14 clusters. The largest clusters had 12 and 17 isolates. The average heterocigosities per state were 21.4% (NL), 15.6% (TAM), (15.6% COA) and 9.9% (DUR). The genetic distances of the isolates between states did not show differences (P > 0.05) when examined by Chi-square tests. The average genetic diversity (15.6%) was due to the variation of strains within subpopulations. In this study an 8.3% difference among states was obtained, which suggest the idea of a unique strain of M. bovis with many variants and that the genetic diversity found for M. bovis could be in part to animal movement between regions. Statistical analysis did not show association (P > 0.05) between risk factors and strains ofM. bovis.
Keywords: Bovine tuberculosis; Spoligotypes; M. bovis; Risk factors.
Resumen
El objetivo de este estudio fue determinar el patrón molecular de aislamientos de M. bovis en ganado del Noreste de México y su asociación con algunos factores de riesgo. Los aislamientos (n=60) fueron obtenidos de los estados de Coahuila (COA, n=14), Tamaulipas (TAM, n=16), Nuevo León (NL, n=14) y Baja California y Durango (DUR, n=16). Los factores de riesgo estudiados fueron: sistema de producción (Ganado para carne o leche), estado, edad, tipo de lesión (localizada y generalizada), y tipo de presentación (caseosa y calcificada). Las muestras fueron analizadas en el laboratorio regional de Monterrey NL, siguiendo un protocolo para espoligotipos. Veinticinco espoligotipos pertenecientes al complejo M. bovis fueron identificados. Once (18.3%) aislamientos presentaron un patrón único, mientras que 49 (81.7%) fueron agrupados en 14 conglomerados. Los conglomerados más grandes tuvieron 12 y 17 aislamientos. Las heterocigosidades promedio por estado fueron 21.4% (NL), 15.6% (TAM), (15.6% COA) y 9.9% (DUR). Las distancias genéticas de los aislamientos entre estados no mostró significancia (P > 0.05), cuando se examino mediante la prueba de Jicuadrada. El promedio de diversidad genética (15.6%) se debió a la variación entre cepas dentro de subpoblaciones. En este estudio se obtuvo un 8.3% de diferencia entre estados, lo que sugiere la idea de una única cepa de M. bovis con muchas variantes y que la diversidad genética encontrada para M. bovis podría ser debida en parte al movimiento de animales entre regiones. El análisis estadístico no mostró asociación (P > 0.05) entre los factores de riesgo y las cepas de M. bovis.
Palabras clave: Tuberculosis bovina; Espoligotipos; M. bovis; Factores de riesgo.
INTRODUCTION
Mycobacterium bovis causes tuberculosis in cattle and wild animals. Bovine tuberculosis (BT) is a disease that affects the mobilization and commercialization of animals and their products, causing millionaire losses due to the impossibility of exporting cattle to the USA (Gurria, 1994). Molecular tracking of M. bovis isolates could help to reinforce epidemiologic studies (Liebana et al, 1997). Spoligotyping technique facilitates the identification and differentiation among M. bovis strains compared to other molecular techniques. Spoligotyping technique has been used to identify sources of infection, transmission between species and stability of the strains in populations during long periods (Aranaz et al, 1996). Genetic diversity has been observed between M. bovis strains in cattle populations, associated to nonlethal mutations that occur in mycobacteria (Cousins et al., 1998) and by the indiscriminate animal mobilization (Kamerbeek et al., 1997; Milian-Suazo et al, 2000). Thirty-five different genetic M. bovis types were found in a study where 273 strains were grouped by region. However, in another study, the same strain was found during 10 years in a milk operation (Cousins et al., 1998). In central Mexico a study with 62 M. bovis isolates using the spoligotyping technique showed certain variability between regions (Milian-Suazo et al, 2000). The objective of this study was to determine the molecular pattern of M. bovis isolates and its association with risk factors in cattle from the Northern region of Mexico.
MATERIAL AND METHODS
Isolates
Sixty isolates from dairy and beef cattle from four states of the North of Mexico: Coahuila (n = 14), Tamaulipas (n = 16), Nuevo Leon (n = 14) and Baja California and Durango (n = 16) were selected for molecular analysis. The isolates were obtained between 2004 and 2006 from animals with lesions compatible with bovine tuberculosis (BT). The isolates were donated by the Regional Laboratory of the Committee for the Foment and Cattle Protection in Nuevo Leon.
The strains of M. bovis were isolated from lymphatic ganglia and organs of suspicious animals of infection with bacteria of the tuberculosis complex specifically M. bovis. From each strain a small sample was taken from the colony and transferred to tubes of 15 ml containing distilled water (1 ml) and 3 or 4 glass pearls, previously sterilized. Then the solution was shaken in a vortex for 10 to 20 seconds to disintegrate the bacterial culture; after that 0.15 ml were taken with a pipette and inoculated into a solid culture medium (Middlebrook and Stonebrink) specific for M. bovis.
At the same time control strains of M. bovis BCG, M. tuberculosis (H37Rv) and a negative control were worked.
DNA extraction
The DNA extraction was made according to Morales et al. (2005) using detergent enzymes. A sample with a loop was taken from the bacterial culture in an Eppendorf tube of 1.5 ml with buffer TE 1X pH 8, inactivated at 80 °C for 30 min and let to cool off; then 50 μl of lysozyme (Amersham Pharmacia Biotech) was added to a concentration of 10 mg/ml, and incubated during one hour. Next 75 μl of sodium dodecil sulphate (SDS) were added at 10% plus 10 μl of K proteinase (Invitrogen) using a concentration of 20 mg/ml. After that the sample was homogenized and incubated at 65 °C for 3 h; 100 μl of NaCl 5 M were added to the dample which was shaken and mixed with 100 μl of CTAB (10 mM Tris-HCl pH=8, 20 mM EDTA, 1.4 M NaCl, 1% PVP and 2% CTAB) previously warmed at 65 °C. Next it was shaken and incubated at 65 °C for 10 min.
The tubes were incubated for 5 min in boiling water to inactivate enzymes and 750 μl of a chloroform solution isoamilic alcohol 24:1 (SEVAG) was added. The solution was homogenized by shaking and centrifuging at 10,000 rpm for 6 min. The supernatant (600 μl approximately) was transferred to a new tube without taking the intermediate phase. Zero point six volumes of isopropanol were added (360 μl approximately) to precipitate nucleic acids. The samples were incubated at 20 °C for 30 min and later were centrifuged at 10,000 rpm for 15 min Most of supernatant was poured off. One milliliter of 70% cold ethanol was added and later centrifuged at 10,000 rpm for 5 min. The supernatant was poured off, air- drying the DNA to room temperature; the DNA was suspended in 20 ul of TE buffer pH=8 at a concentration of 1X and incubated 30 min at 65 °C. Finally the sample was stored at -20 °C until its use.
Molecular analysis protocol
Spoligotyping was carried out following the protocol described by Kamerbeek et al. (1997). Briefly, DNA was amplified with AmpliTaq DNA Polymerase (Bioline, Inc.) in a 50 μL PCR mix, containing 5 μl of lOx reaction buffer (160 mM (NH4)2SO4, 670 mM Tris-HCl (pH 8.8 at 25°C), 0.1% stabilize)-, 1.5 mM of MgCl2, 0.2 mM of each dNTP, 25 pmol of each oligonucleotide primer (DRa 5 - GGT TTT GGG TCT GAC GAC -3' marked with biotin at 5'-end, and DRb 5' - CCG AGA GGG GAC GGA AAC-3) and 5 μl, (100-200 ng) of template DNA. The mixture was heated in a PCR Express (ThermoHybaid, UK) at 96 °C for 3 min and subjected to 35 cycles of denaturation at 96 °C for 1 min, annealing at 55 °C for 1 min and extension at 72 °C for 35 sec followed by a final extension of 72 °C for 5 min.
The amplified DNA was visualized through electrophoresis in a SDS-polyacrylamide 6% gel stained with ethidium bromide, at concentration of 10 mg/ml, at 100 V for 90 min. Twenty μl of PCR product was added to 150 μl of 2X SSPE-0.1% SDS and heat denatured at 99 °C for 10 min, and applied to a nylon membrane to which 37 spacers sequences from M. tuberculosis and six spacer sequences for M. bovis BCG were covalently bound (ISOGEN, Maarssen, The Netherlands). This membrane was then placed in a miniblotter MN45 (Immunetics; Cambridge, MA) in such a way that the slots were perpendicular to the line pattern of the previously applied oligonucleotides. The membranes were then incubated in 10 ml of 2X SSPE-0.5% SDS plus 5 μl of streptavidin-peroxidase conjugate for 45 min at 42 °C. Hybridized DNA was detected with the ECL detection liquid (Amersham Biosciences; Piscataway, NJ); and by exposing the ECL-film (Kodak Istman) to the membrane for 12 min. The control strains were included in the molecular analysis.
Risk factors
The risk factors evaluated were: production system (meat and milk), state (COA, TAM, NL and DUR), group of age (1-2, 3-4, 5-6 and 7-15 years), type of presentation (localized and generalized) and type of lesion (caseous and calcified). All the factors were registered in the field sheets of the TIF sent with the samples to the Central laboratory. Each isolated strain was related to some factor through the count of bands obtained from the membrane of 37 spacers of sequences for M. tuberculosis and six spacers of sequences for M. bovis. These factors were later analyzed to know the epidemiological association between the factors and the molecular patterns of the cattle isolates of Northern Mexico.
Statistical analysis
Marking as 1 the presence and 0 the absence of bands in the molecular patterns of the isolates, a matrix of 0' s and to 1' s was built in Excel which was used in the analysis of comparison of pairs of isolates to obtain the matrix of genetic distances (Nei and Li, 1979). The distances were used in a cluster analysis using the unweighted-pairwise-group method average (UPGMA) to obtain the dendogram showing the molecular tree. Also, the dendogram was analyzed to determine if the related isolates were epidemiologically grouped.
The genetic diversity between subpopulations or categories of the risk factors was measured using the formula: GST = DST/HT; where: GST measures the proportion of the genetic diversity due to differences between subpopulations; DST is the genetic diversity between subpopulations; HT is the total genetic diversity in the 60 isolates = Hs + DST; and HS it is the genetic diversity within subpopulations. All the statistical analyses were made with the POPGENE program (Yeh et al, 1999).
Finally, the samples were classified as grouped, when they were isolated and clustered by UPGMA method, or not grouped when the strain was isolated but did not belong to any cluster or it was considered as a unique isolate. To determine the association of risk factors with grouped or non-grouped isolates, the exact Fisher test was used (SAS, 2002).
RESULTS
Molecular typing
The dendogram with the population structure of the isolates of M. bovis is shown in Figure 1. Twenty-five patterns of spoligotypes were obtained from the 60 isolates; eleven (18.3%) isolates were represented by a unique pattern, whereas 49 (81.7%) were grouped in 14 conglomerates. The largest conglomerates had 12 and 7 isolates.
Diversity and genetic distance
Total genetic diversity (HT =15.6%) could be attributed to the variation of the strains within the subpopulations, since only one small contribution of the total variation (1.14 to 8.30%) came from difference between subpopulations.
The genetic differences between isolates classified by risk factor such as production system, region, age, presence of disease and type of lesion are shown in Table 1. The average heterocigosity between the subpopulations varied from 9.9% in the DUR subpopulation to 21.4% in the NL subpopulation. Thirty-five of the 43 markers (81.4%) used were polymorphic. The genetic distances were small and no significant. The greatest variation between subpopulations (GST=8.3%) corresponded to the difference between states (Table 1); which indicates that only 8.3% of the total variation is explained by the difference of strains between regions and 91.7% by the variation of strains within regions. Figure 1 shows that the strains of a region or state could have different genotypes, whereas some of the regions present a similar genetic pattern.
Risk factors
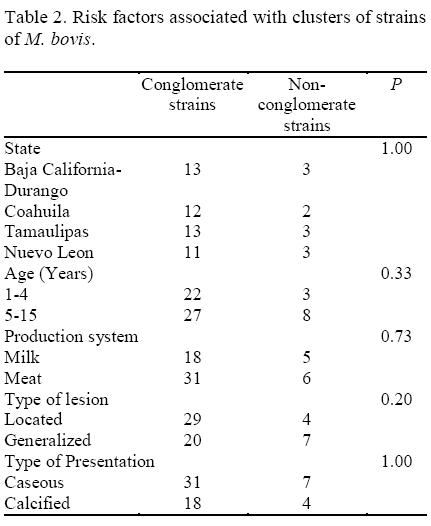
The results of the molecular analysis of M. bovis for the subpopulations of isolates are presented in Table 2. The statistical analysis of the data did not show significant association between the risk factors investigated and grouped and non grouped strains.
DISCUSSION
Molecular typing of M. bovis
The dendogram of Figure 1 shows a wide genetic variability between isolates, since 81.4% of the strains were polymorphic. In other studies, genetic diversity (heterocigosity) of M. bovis has been related to the populations throughout the indiscriminate movement of cattle between the regions of a country (Milian-Suazo et al., 2000). However, this would be possible if differences of strains between regions existed, which would affect their frequencies, due to infections of local animals by strains of imported animals. The results of this study do not support this hypothesis. On the contrary they suggest that samples belong to a clone population (unique strain) with wide genetic diversity, as has been suggested by Vázquez-Marrufo et al. (2008). Therefore, the similar genetic patterns of the isolates suggest a common source of infection (Salamon et al., 2000). The method of typing and the number of fragments included in the statistical analysis may affect the similarity of the fingerprinting (Van Embden et al., 1993).
Diversity and genetic distance
The analyses of the genetic distances between isolates from each category of the risk factors (production system, region, group of age, etc.) did not show statistical differences between strains. This supports the idea that the genetic diversity of M. bovis it probably due to the indiscriminate movement of animals between regions of a country (Perumaalla et al., 1996) and to the form in that the bacterium is disseminated. The theory of the molecular typing establishes that epidemiologically related isolates are derived from the clone expansion of a single precursor and, as a result, have common characteristics that differ from those that are unrelated epidemiologically (Maslow and Mulligan, 1993). The use of a particular characteristic for typing is related to its stability within a strain and its diversity within the species; therefore, it is expected that strains of the same region are more related than strains from different regions. Nevertheless, those studies and one in Central Mexico (Milian-Suazo et al., 2002) show that this it is not the case for the M. bovis strains in Northern Mexico.
Risk factors
In this study was not possible to identify any pattern based on the strains categorized as grouped and not grouped. However, it has been shown that the genetic diversity of M. bovis strains is present even though animal mixture of different origin does not exist, which differs from previous reports (Perumaalla et al., 1996). In goats, there is not significant differences in the distributions by sex and age, although there were marked differences by geographic distribution (Kubica et al., 2003). It was expected that age was an important risk factor but our results do not support this. This probably was due to the culling process of animals, which maintains prevalences similar between age groups. Conversely, in countries like Great Britain and Ireland (Hewinson et al., 2006) geographic distribution of spoligotypes has been observed. In Mexico, an association between the patterns of spoligotypes and geographic location has not been notified.
CONCLUSION
The isolates classified by region, group of age, production system, type of lesion and type of presentation did not show independence which suggests a common origin of the M. bovis strains. Although the diversity of M. bovis isolates is wide, it does not follow a geographic pattern. It was not possible to identify risk factors based on the strains being categorized as grouped and not grouped.
ACKNOWLEDGEMENT
Isaac Padilla thanks the Consejo Nacional de Ciencia y Tecnología (CONACYT) for the scholarship provided during his master science studies.
REFERENCES
Aranaz, A., Lebiana, E., Mateos, A., Dominguez, L., Vidal, D., Domingo, M., Gonzalez, O., Rodriguez-Ferri, E.F., Bunschoten, A.E., Van Embden, J.D., Cousins, D. 1996. Spacer oligonucleotide typing of Mycobacterium bovis strains from cattle and animals: a tool for studying epidemiology of Tuberculosis. Journal of Clinical Microbiology 34:2734-2740. [ Links ]
Cousins, D., Williams, S., Liébana, E., Aranaz, A., Bunschoten, A., Van Embden, I, Ellis, T. 1998. Evaluation of four DNA typing techniques in epidemiological investigations of bovine tuberculosis. Journal of Clinical Microbiology 3:168-178. [ Links ]
Gurria, T.F. 1994. Situación actual de la campaña contra la tuberculosis y brucelosis en México. México Ganadero 385:21-28. [ Links ]
Hewinson, R.G., Vordermeier, H.M., Smith, N, Gordon, S.V 2006. Recent advances in our knowledge of Mycobacterium bovis: a feeling for the organism. Veterinary Microbiology 112:127-139. [ Links ]
Kamerbeek, J., Schouls, L., Kolk, A., Van Agterveld, M., Van Soolingen, D., Kuiper, S., Bunschoten, A., Molhuizen, H., Shaw, R., Goyal, M., Van Embden, J. 1997. Simultaneous strain detection and differentiation of Mycobacterium tuberculosis for diagnosis and epidemiology. Journal of Clinical Microbiology 35, 907- 914. [ Links ]
Kubica, T., Rusch-Gerdes, S., Niemann, S. 2003. Mycobacterium bovis subsp. Caprae caused one-third of human Mycobacterium bovis -associate tuberculosis cases reported in Germany between 1999 and 2001. Journal of Clinical Microbiology 41:3070-3077. [ Links ]
Liebana, E., Aranaz, A., Dominguez, L., Mateo, A., Gonzalez, O. 1997. The insertion element IS6110 is a useful tool for DNA fingerpringting of M. bovis isolates from de cattle and goats in Spain. Veterinary Microbiology 54:223-233. [ Links ]
Maslow, J., Mulligan, M. 1993. Molecular epidemiology: application of contemporary techniques to the typing of microorganisms. Clinical Infectious Disease. 17, 153-164. [ Links ]
Milian-Suazo, F., Salman, M., William, C.B., Triantis, J., Ramirez, C, Payeur, J., Torres, M. 2000. Molecular epidemiologic analysis of Mycobacterium bovis isolates from Mexico. American Journal of Veterinary Research 61:90-95. [ Links ]
Milian-Suazo, F., Banda-Ruiz, V, Ramirez, C.C., Amaga, D.C. 2002. Genotyping of Mycobacterium bovis by geographic location within Mexico. Preventive Veterinary Medicine 55:255-264. [ Links ]
Morales, A., Martinez, I., Carlos, A., Alvarez, G., Alvarez, M., Maldonado, J. 2005. Comparación de histopatología, cultivo y PCR en el diagnóstico de tuberculosis bovina. Revista Científica FCV-LUZ 15:103-108. [ Links ]
Nei, M., Li, W.H. 1979. Mathematical model for studying genetic variation in terms of restriction endonucleases. Proceedings of the National Academy of Science 74:5267-5273. [ Links ]
Perumaalla, V.S., Adams, L.G., Payeur, J.B., Jarnagin, J.J., Baca, D.R., Quemes, F.S., Fitch, T.A. 1996. Molecular epidemiology of Mycobacterium bovis in Texas and México. Journal of Clinical Microbiology 34:2066-2071. [ Links ]
Salamon, H., Behr, M.A., Rhee, J.T., Small, P.M. 2000. Genetic distances for the study of infectious diseases epidemiology. American Journal of Epidemiology 151:324-334. [ Links ]
SAS. 2002. SAS/STAT User's Guide, Ver. 9.0. SAS Institute Inc, Cary, North Carolina, USA. [ Links ]
Van Embden, ID., Cave, M.D., Crawford, J.T., Dale, J.W., Eisenach, K.D., Gicquel, B., Hermans, P., Martin, C, McAdam, R., Shinnick, T.M. 1993. Strain identification of Mycobacterium tuberculosis by DNA fingerprinting: recommendations for a standardized methodology. Journal of Clinical Microbiology 31:406-409. [ Links ]
Vázquez-Marrufo, G., Marín-Hernández, D., Zavala-Páramo, M.G., Vázquez-Narváez, G., Álvarez-Aguilar, C, Vázquez-Garcidueñas, M.S. 2008. Genetic diversity among Mycobacterium tuberculosis isolates from mexican patients. Canadian Journal of Microbiology 54:610-618. [ Links ]
Yeh, F.C., Yang, R., Boyle, T. 1999. POPGENE version 1.31. Microsoft Window-based freeware for Population Genetic Analysis. University of Alberta, Edmonton, Alberta, Canada. http://www.alberta.ca/fyeh/popgene.pdf [ Links ]

