










Servicios Personalizados
Revista

Articulo
 Español (pdf)
Español (pdf)
 Artículo en XML
Artículo en XML Referencias del artículo
Referencias del artículo
 Enviar artículo por email
Enviar artículo por emailIndicadores
 Citado por SciELO
Citado por SciELO Links relacionados
 Similares en
SciELO
Similares en
SciELO Compartir

 Permalink
PermalinkRevista mexicana de ciencias farmacéuticas
versión impresa ISSN 1870-0195
Rev. mex. cienc. farm vol.43 no.2 Ciudad de México abr./jun. 2012
Revisión bibliográfica
Colesterol: Función biológica e implicaciones médicas
Cholesterol: Biological function and medical implications
Octavio Maldonado Saavedra,1 Israel Ramírez Sánchez,2 José Rubén García Sánchez,2 Guillermo Manuel Ceballos Reyes,2 Enrique Méndez Bolaina1,3
1 CIB-Doctorado en Ciencias Biomédicas-UV.
2 SEPI-Escuela Superior de Medicina-IPN.
3 Facultad de Ciencias Químicas-Universidad Veracruzana.
Correspondencia:
Dr. Enrique Méndez Bolaina.
Facultad de Ciencias Químicas Universidad Veracruzana.
Prolongación de Oriente 6 # 1009
Colonia Rafael Alvarado, Orizaba, Veracruz
C.P. 94340, A. P. 215, México
Tel: (272) 7240120, Fax: 7241779
e-mail: enmendez@uv.mx
Fecha de recepción: 17 de agosto de 2011.
Fecha de recepción de modificaciones: 1 de marzo de 2012.
Fecha de aceptación: 5 de marzo de 2012.
Resumen
Nuestro organismo sintetiza su propio colesterol, un porcentaje extra es obtenido de la dieta. El colesterol forma parte de las membranas de nuestras células, sirve como precursor de todas las hormonas esteroideas, ácidos biliares, y de la vitamina D. La ingesta excesiva de colesterol y las mutaciones genéticas en el rLDL promueven el incremento de colesterol sérico por encima de los niveles recomendables, lo que favorece la génesis de la ateroesclerosis, la cual, es el principal factor riesgo para el desarrollo de enfermedades cardiovasculares. Estilos de vida saludables, tratamientos dietéticos y farmacológicos logran reducir las concentraciones de colesterol plasmático, lo que disminuye la probabilidad de sufrir un evento coronario.
En este artículo se revisarán el papel biológico del colesterol, su relación con las enfermedades cardiovasculares, además analizaremos aspectos dietéticos y farmacológicos que ayudan a contrarrestar estos padecimientos.
Palabras clave: colesterol, hipercolesterolemia, disfunción endotelial, ateroesclerosis, enfermedades cardiovasculares.
Abstract
Our body synthesizes its own cholesterol, an extra percentage is obtained from diet. Cholesterol is part of our cell membranes, serves as a precursor of all steroid hormones, bile acids, and vitamin D. Excessive intake of cholesterol and genetic mutations in the rLDL promotes the increase of blood serum cholesterol above recommended levels, which facilitate the genesis of atherosclerosis, that is the main risk factor for developing cardiovascular disease. Healthy lifestyles, dietary and pharmacological treatments are able to reduce plasma cholesterol concentrations, which decreases the probability to suffer a coronary event.
Here, we review the biological role of cholesterol, its relationship with cardiovascular disease also discusses dietary and pharmacological aspects help to counteract these conditions.
Key words: cholesterol, hypercholesterolemia, endothelial dysfunction, atherosclerosis, cardiovascular disease.
Introducción
El colesterol (3-hidroxi-5,6 colesteno) es una molécula indispensable para la vida, desempeña funciones estructurales y metabólicas que son vitales para el ser humano. Se encuentra anclado estratégicamente en las membranas de cada célula donde modula la fluidez, permeabilidad y en consecuencia su función.1 Esta regulación implica que el contenido en colesterol de las membranas modifica la actividad de las enzimas ancladas en ellas, así como la de algunas proteínas transportadoras y de receptores de membrana.2 El colesterol proviene de la dieta o es sintetizado por nuestras células (principalmente en los hepatocitos); es precursor de otras biomoléculas fisiológicamente importantes tales como, las hormonas esteroideas (andrógenos, estrógenos, progestágenos, gluco y mineralcorticoides), ácidos biliares y la vitamina D.3,4 Sin embargo, la acumulación excesiva de colesterol en nuestros tejidos y altas concentraciones en sangre (hipercolesterolemia), pueden tener consecuencias patológicas altamente prevalentes en la población Mexicana. Esto es particularmente cierto para las células endoteliales que forman la pared arterial, donde la acumulación de colesterol inicia la enfermedad cardiovascular aterosclerótica.5 Numerosos estudios epidemiológicos y retrospectivos han mostrado una relación directa entre el colesterol total y el colesterol unido a Lipoproteínas de Baja Densidad (C-LDL) con la morbilidad y mortalidad debida a causas cardiovasculares.6,7 El efecto concomitante en la inhibición de la enzima 3-hidroxi-3-metilglutaril-coenzima A reductasa (HMG-CoAR) y de la absorción de colesterol a nivel intestinal, mediante estatinas y ezetimiba, respectivamente, maximizan el descenso del C-LDL, relacionado con una reducción de las complicaciones cardiovasculares como la hipertensión, la ateroesclerosis y muerte por complicaciones coronarias.8,9
El objetivo de esta revisión es la recopilación y el aporte de un compendio de información amplia, relevante y actualizada que aborda el importante papel que juega el colesterol en nuestro organismo, así como la relación del curso patológico que se presenta (hipercolesterolemia, ateroesclerosis e infarto) por la acumulación excesiva de colesterol en la pared endotelial, producto de la ingesta de dietas ricas en grasa o por un carácter de índole genético, conocido como Hipercolesterolemia Familiar. Se analizarán aspectos dietéticos y farmacológicos que ayudan a disminuir los niveles de colesterol plasmático.
El colesterol (ver Figura 1) presente en nuestro organismo, se obtiene principalmente de dos fuentes: De la dieta (colesterol exógeno) y la síntesis endógena (colesterol endógeno). Prácticamente todos los tejidos que contienen células nucleadas son capaces de sintetizar colesterol. La fracción microsómica (retículo endoplásmico) del citosol es responsable de susíntesis.10
La tasa de síntesis de colesterol es regulada por la HMG-CoAR,11 cuya actividad es controlada por el flujo de colesterol intestinal hacia el hígado. La acetil-CoA proporciona todos los átomos de carbono para la síntesis del colesterol, la cual puede dividirse en cinco etapas:
1. Síntesis de mevalonato, un compuesto de seis carbonos, a partir de acetil-CoA.
2. Formación de unidades isoprenoides por pérdida de CO2 del mevalonato.
3. Condensación de seis unidades isoprenoides para formar el intermediario, escualeno.
4. Cierre del escualeno para la formación cíclica del esteroide precursor, conocido como lanosterol.
5. El colesterol se forma del lanosterol después de varios pasos posteriores que incluyen la pérdida de tres grupos metilo.12,13
Absorción intestinal de colesterol
La absorción de colesterol en el intestino delgado proximal representa la principal vía de entrada del colesterol hacia nuestro cuerpo. Los factores que influyen sobre la absorción de colesterol son múltiples, entre los más importantes destacan, la edad, la cantidad y la composición de los ácidos biliares, los factores dietéticos y genéticos, además de la composición y densidad bacteriana que existe en la flora intestinal. El colesterol presente en la luz intestinal deriva principalmente de la secreción biliar, la ingesta alimentaria y en menor proporción de la descamación del epitelio intestinal. La absorción del colesterol intestinal es un proceso complejo, que incluye por lo menos 3 fases: 1) Intraluminal (solubilización micelar), 2) mucosa (de transporte a través de la membrana apical para la absorción en los enterocitos), 3) intracelular (movilización de los quilomicrones y su secreción a la linfa y sangre a través de la membrana basolateral de los enterocitos).14
Fase intraluminal
El colesterol al igual que los triglicéridos es poco soluble en sistemas acuosos. Lo anterior justifica la dependencia absoluta de la absorción del colesterol en la capacidad emulsificante de los ácidos biliares. Los ácidos biliares son moléculas anfipáticas derivadas del colesterol, capaces de solubilizar lípidos cuando se agrupan en una cantidad mayor a la concentración micelar crítica (concentración mínima de surfactante en este caso ácidos biliares, a partir de la cual se forman micelas espontáneamente en una disolución). La síntesis de ácidos biliares genera el flujo de bilis desde el hígado hasta el intestino.15 El transporte de los ácidos biliares entre el hígado y el intestino se conoce como la circulación enterohepática de la bilis. Los ácidos biliares son detergentes biológicos que facilitan la excreción biliar de los metabolitos del colesterol endógeno y sustancias ajenas al organismo, así también facilitan la absorción intestinal de los principales lípidos de la dieta (grasas o aceites, también llamados triglicéridos, fosfolípidos, ésteres de colesterol) y nutrientes. El hígado juega un papel central en el mantenimiento de la homeostasis del colesterol mediante el equilibrio de múltiples vías, incluyendo la síntesis de colesterol endógeno, así como la de los ácidos biliares, la absorción de colesterol dela dieta, la excreción biliar de colesterol, la síntesis de las lipoproteínas, y el transporte reverso del colesterol.16
La formación de las micelas es requisito indispensable para la absorción de colesterol. Tras la ingesta de alimentos, se estimula la contracción de la vesícula biliar desencadenado la liberación de un gran volumen de sales biliares. En la porción proximal del intestino delgado, los lípidos y sales biliares interactúan espontáneamente y forman las micelas mixtas (principalmente fosfolípidos y colesterol no esterificado). La solubilización micelar constituye un mecanismo de transporte para que el colesterol, logre difundirse a través de la barrera mucosa que recubre la superficie de las microvellosidades intestinales, donde las micelas terminan su función de trasporte y se disgregan, tras lo cual los monómeros de colesterol están disponibles para ser internalizados en los enterocitos, las células del epitelio intestinal.17,18
Fase mucosa
En esta fase el colesterol presente en el lumen pasa al citoplasma de los enterocitos a través del borde en cepillo. Durante mucho tiempo el proceso de absorción de colesterol se había considerado el resultado de un balance entre la difusión pasiva de entrada a través de la membrana del enterocito y la excreción de vuelta a la luz intestinal.19
-Internalizacion de colesterol al enterocito
Niemann- Pick C1-like protein 1 (NPC1L1) es una proteína que se expresa en el borde en cepillo de los enterocitos del intestino delgado, el cual transporta el colesterol dietético y biliar desde el lumen intestinal al enterocito para facilitar la absorción del colesterol. Deleciones de NPC1L1 en ratones produce una dramática reducción de la absorción de colesterol. En el hígado, NPC1L1 se localiza en la membrana canalicular biliar. Se encontró en el ser humano NPC1L1, cuando transgénicamente se sobreexpresó en el hígado del ratón, se localizó en la membrana canalicular biliar. En estos animales transgénicos, los niveles de colesterol en la bilis se reducen drásticamente, sin efectos sobre la expresión hepática de los transportadores de eflujo del colesterol ABCG5/G8. Este hallazgo apoya la idea de que la NPC1L1 hepática puede contrarrestar la secreción biliar de esteroles mediada por ABCG5/G8, mediante el transporte de esteroles biliares de nuevo a los hepatocitos.20 Este mecanismo puede haber evolucionado para proteger contra la pérdida excesiva colesterol biliar, un componente esencial estructural de la membrana celular.21,22
-Exportación del colesterol desde los enterocitos hacia el lumen intestinal
Por otro lado, se conoce que dos transportadores de membrana ABCG5 y ABCG8 actúan como heterodímeros, encargados de devolver los esteroles absorbidos al lumen intestinal. El descubrimiento de que mutaciones en los genes ABCG5 y ABCG8, eran causantes de la β-sitosterolemia, enfermedad autosómica recesiva debida a hiperabsorción intestinal de esteroles y caracterizada por la aparición de xantomatosis y ateroesclerosis prematura, dio relevancia a su estudio.23,24
Los genes ABCG5 y ABCG8 se encuentran regulados por el colesterol, de modo que, cuando aumenta la ingesta de este lípido aumenta la expresión intestinal y hepática de estos genes. Probablemente, su función es responder a la sobrecarga de esteroles y devolverlos hacia la bilis o hacia el intestino e impedir de esta forma que el organismo sufra las consecuencias de una sobrecarga de colesterol. El hecho de que los dos transportadores opuestos de esteroles, NPC1L1 y ABCG5/G8, evolucionaron en la misma localización subcelular en los enterocitos y hepatocitos destaca la importancia del equilibrio de esteroles en la vida.25
Fase intracelular
Aproximadamente la mitad del colesterol que ha sido captado por los enterocitos y no ha sido devuelto al lumen intestinal por la vía ABCG5/8 se difunde al retículo endoplasmático, donde es reesterificado por la enzima Acil-CoA: Colesterol Aciltransferasa-2 (ACAT2), presente en el intestino y en el hígado fetal, cumpliendo la misma función de la ACAT1 presente en el hígado, glándula suprarrenal, los macrófagos y el riñón.7,26 Generalmente se esterifica con ácido palmítico, aunque si la disponibilidad del ácido oleico dietario es alta, también se utiliza este ácido.
Homeostasis del colesterol
El colesterol es sintetizado principalmente en el hígado a través de una amplia serie de reacciones. En la Figura 2 se esquematizan las interacciones entre los tejidos periféricos, el hígado y el intestino en el mantenimiento de la homeostasis del colesterol. El hígado representa un papel central en la regulación del metabolismo del colesterol y de las cifras séricas de C-LDL. En situaciones de equilibrio homeostático, la cantidad de colesterol excretada diariamente en las heces (unos 1 100 mg, procedentes de la dieta, la bilis y la descamación epitelial intestinal) es igual a la suma del sintetizado por los tejidos (unos 800 mg) y del aportado por las comidas (unos 300 mg).30
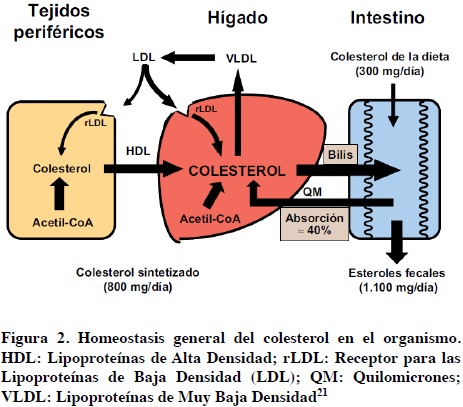
El equilibrio del colesterol es regulado por mecanismos de retroalimentación entre las vías endógena y exógena del metabolismo del colesterol. Una reducción de la entrada de colesterol intestinal por inhibición de su absorción aumenta la actividad de la HMG-CoAR e intensifica la síntesis de colesterol. En cambio, una captación intestinal elevada de colesterol inhibe la HMG-CoAR, reduce la síntesis hepática y produce una regulación a la baja de los rLDL, por medio de la reducción de la captación de LDL y el aumento sus concentraciones plasmáticas.31
Por el contrario, cuando existe una depleción de colesterol, los rLDL son regulados a la alza y dan lugar a un aumento de la eliminación de partículas de LDL de la sangre así como a una disminución de las concentraciones plasmáticas de C-LDL. Este equilibrio determina que para obtener una eficacia máxima en la reducción del colesterol sea necesario un doble mecanismo de acción, por un lado inhibiendo la síntesis de colesterol mediante bloqueo de la HMG-CoAR y, por otro, bloqueando la absorción del colesterol intestinal.31,32
Transporte de colesterol: lipoproteínas
Los lípidos plasmáticos (triglicéridos, ésteres del colesterol, fosfolípidos y colesterol libre), son insolubles en medios acuosos como la sangre, de tal forma que deben ser empaquetados en partículas de lipoproteína para su transporte. Cerca del 70% del colesterol se encuentra unido a lipoproteínas plasmáticas en forma de ésteres de colesterol. Las lipoproteínas son ensamblados macromoleculares de proteínas y lípidos.33
Estructuralmente consisten en un núcleo de lípidos apolares (colesterol esterificado y triglicéridos) rodeado por una capa exterior donde residen proteínas denominadas apolipoproteínas (apo), lípidos anfipáticos (con dos porciones, una polar y otra apolar) con su parte polar hacia la parte exterior de la partícula. Cada lipoproteína contiene una o más apolipoproteínas, que proporcionan estabilidad estructural, además de servir como ligandos para receptores celulares que determinan el destino metabólico de partículas individuales o actúan como cofactores para enzimas comprendidas en el metabolismo de las lipoproteínas.34
Las lipoproteínas cuentan con distintas densidades, esto se debe a la proporción relativa de lípidos y proteínas que posee cada partícula.
Dentro de las lipoproteínas de mayor importancia clínica encontramos las de alta densidad (HDL, del inglés, High-Density Lipoprotein), de baja densidad (LDL, del inglés, Low-Density Lipoprotein), de muy baja densidad (VLDL, del inglés, Very Low-Density Lipoprotein), de densidad intermedia (IDL, del inglés, Intermediate-Density Lipoprotein), la lipoproteína (a) [Lp(a)] y los quilomicrones (QM).
Los quilomicrones son partículas lipoproteicas de gran tamaño que se sintetizan en los enterocitos, están constituidos por ~90% triglicéridos, 7% de fosfolípidos, 1% colesterol, y un 2% de proteínas especializadas, mayoritariamente apo B-48. Esta apoproteína pertenece a la misma familia de la B-100, de hecho, la apo B-48 y la apo B-100 están codificadas por el mismo gen, solo que la apo B-48 contiene el 48% de la longitud total de la apo B-100 y se expresan en lugares diferentes. La apo B-48 se sintetiza en el intestino delgado, le confiere soporte estructural a los quilomicrones y permite su secreción desde el hígado. La apo B-100 es sintetizada en el hígado, y se encuentra en las VLDL, IDL y HDL.35
Los lípidos (vía exógena) de la dieta son absorbidos en el intestino por los quilomicrones, que son secretadas en la linfa y pasan a la sangre a través del conducto torácico. Posteriormente los quilomicrones, se someten a la lipólisis rápida por parte de la Lipoproteína Lipasa (LPL) en los lechos capilares extrahepáticos, un proceso que elimina algunos de los TG y deja pequeños remanentes de quilomicrones que internalizan el resto de los lípidos de la dieta al hígado. En la vía endógena, el hígado utiliza los remanentes de quilomicrones, lípidos y colesterol endógeno para producir las partículas de VLDL.36,37
Por otro lado, es importante mencionar, que erróneamente se había creído durante muchos años, que el tamaño de los remanentes de quilomicrones impedía su entrada a la pared arterial y su posterior interacción con los macrófagos. Sin embargo, ahora está claro que los remanentes de quilomicrones puede penetrar y quedar retenidos en el espacio subendotelial tan eficientemente como las Lipoproteínas de Baja Densidad (LDL). La presencia de apo B-48 aislada de las placas ateroscleróticas y de restos de quilomicrones en macrófagos y células espumosas, evidencian la desconocida aterogenicidad de esta lipoproteína.38
Las VLDL son lipoproteínas ricas en triglicéridos de origen endógeno, se sintetizan en el hígado. Contienen entre 10-15% del colesterol plasmático, fosfolípidos y un conjunto característico de apolipoproteínas: Apo B-100, apo C-I, apo C-II, apo C-III y apo E. Estas lipoproteínas son transportadas por la sangre desde el hígado hasta el músculo y el tejido adiposo, donde la LPL se activa gracias a la apo C-II, hidrolizando los triglicéridos de las VLDL, liberando ácidos grasos libres que pueden ser almacenados por los adipositos. La pérdida de triglicéridos y algunas de sus apolipoproteínas, convierte a las VLDL en LDL.39 Las C-LDL son sintetizadas en el hígado: Tienen una concentración alta de colesterol y moderada de fosfolípidos, y no contienen triglicéridos. Su apolipoproteína asociada de mayor importancia es apo B-100, indispensable para unirse a su rLDL. Nuestro organismo cuenta con receptores específicos para las LDL en casi todas las membranas celulares, que identifica, capta e interioriza las LDL. Debido a su alta aterogenicidad, es de gran interés clínico, típicamente representa entre 60-70% del colesterol sérico total y su función es transportar el colesterol desde el hígado hacia los tejidos periféricos.40
El C-HDL es conocido por ser protector contra las enfermedades cardiovasculares, extrae colesterol de las lesiones ateroscleróticas y lo transporta hasta el hígado para su posterior metabolismo y eliminación intestinal junto con las heces. El C-HDL se produce en el hígado y en el intestino. La principal proteína de las HDL es la apo A-I (peso molecular 28 000 Da), encargada del destino de las HDL. La apo A-I constituye más del 70% del contenido proteínico del total de partículas de HDL. La apoA-II (peso molecular 17 000 Da) es la segunda apolipoproteína más abundante en las HDL, pero su papel no ha sido bien definido. Otras proteínas que se encuentran en menores cantidades incluyen a las apo C-I, apo C-II, apo C-III, y apo-E. En el plasma, el C-HDL se convierte en un éster de colesterol por acción de la Éster de Colesterol Transferasa (LCAT). Mientras que circulan en el torrente sanguíneo, las partículas de C-HDL adquieren más colesterol del torrente sanguíneo. Además, las partículas de C-HDL van a eliminar el colesterol a través de un proceso de Transporte Inverso de Colesterol (TIC) desde los tejidos periféricos y de ateroma en las arterias hasta el hígado, llevando aproximadamente el 30% del colesterol sérico.41,42,43 Las IDL tienen altas concentraciones de colesterol y fosfolípidos, pues provienen del proceso de degradación de las VLDL, la hidrólisis de los TG libera ácidos grasos y las partículas resultantes de IDL contienen menor porcentaje de TG y fosfolípidos y la misma magnitud de colesterol. Por lo tanto son partículas más heterogéneas, porque la LPL continúa catalizando la degradación de TG y produciendo ácidos grasos libres y glicerol, en tanto esté ligada a la VLDL por la Apo C-II. El final de esta degradación se produce cuando queda muy poca apoC-II y se inhibe por efecto de apo C-III.44
La apo C-II está formado 79 aminoácidos (PM 8 500 Da) es un activador fisiológico de la LPL. Juega un papel muy importante en la regulación del metabolismo de los triglicéridos asociados a quilomicrones y VLDL.45 La apo C-III está formado por 79 aminoácidos (PM 8 800 Da) se sintetiza en hígado e intestino y forma parte de los quilomicrones, VLDL, LDL y HDL y está presente en plasma en su forma glicosilada. Esta apo juega un papel importante en el control del metabolismo y en la concentración plasmática de las lipoproteínas ricas en triglicéridos (VLDL y quilomicrones) y sus remanentes, debido a que inhibe la actividad de la LPL y la lipasa hepática apo C-II y C-III participan en la regulación de la lipasa lipoproteica, generando un efecto de inhibición sobre ella.46
La lipoproteína (a) [Lp(a)], contiene una gran cantidad de ésteres de colesterol y fosfolípidos, su composición es muy similar a la LDL, pero la diferencia esencial entre ambas radica en que la Lp(a) presenta una apo que está unida a la apo B-100 por un puente disulfuro, y es estructuralmente parecida al plasminógeno, esta similitud estructural le confiere la capacidad de unirse con la fibrina y a las proteínas de las membranas celulares. La Lp(a) puede interferir con la fibrinólisis, así como favorecer los depósitos de lípidos y estimular el crecimiento de células musculares lisas, lo cual favorece la aterogénesis, por lo que constituye un factor genético de riesgo para la ateroesclerosis.47,48
Ateroesclerosis y colesterol sérico: disrupción endotelial
La relación entre los altos niveles de colesterol, la ateroesclerosis y la disfunción endotelial ha sido ampliamente estudiada.49,50 Las concentraciones séricas elevadas de Colesterol Total (CT), C-LDL y C-VLDL y concentraciones séricas bajas de C-HDL se correlacionan con la extensión de estas lesiones ateroscleróticas. Las primeras lesiones ateroscleróticas, son la estrías grasas, las cuales pueden ya manifestarse en la aorta y en la arterias coronarias durante las primeras décadas de vida y consisten básicamente en una acumulación de grasas de tipo LDL en la íntima vascular debido a la disfunción endotelial.51
En condiciones fisiológicas, las lipoproteínas que penetran en el espacio subendotelial se devuelven a la sangre circulante por el mecanismo TIC, en el cual participan las HDL. Cuando se produce disfunción endotelial, el aumento de la permeabilidad de la pared de los vasos origina un aumento en la penetración de las LDL en la pared vascular, que excede la velocidad y la eficiencia del sistema de TIC para devolverlo al torrente sanguíneo. Unido a esto, algunos factores de riesgo como la diabetes y el hábito de fumar reducen la cantidad de HDL y disminuyen aún más la eliminación de las LDL.52 Todos estos hechos originan un aumento en el período en que permanecen las lipoproteínas dentro del espacio subendotelial, donde se someten a una oxidación leve, principalmente por las células endoteliales, lo que produce moléculas de LDL Mínimamente Modificadas (MM-LDL) que junto al estrés oxidante presente en el ambiente, la presencia de angiotensina II y la reducción de la presión del flujo, llamada "rozamiento" en las zonas con propensión a la ateroesclerosis, son capaces de activar el Factor Nuclear kappa-β (NF-κβ), factor de transcripción que aumenta la expresión de moléculas que participan en los pasos de captación de monocitos. Dichas moléculas se pueden dividir en 2 grupos:
1. Moléculas de adhesión (VCAM-1, ICAM-1, selectina E):Responsables del movimiento y de la adhesión de monocitosa la pared de los vasos sanguíneos.
2. Moléculas quimioafines (MCP-I, IL-8): Que provocan la entrada de monocitos en la pared de los vasos sanguíneos.39
Una vez en el espacio subendotelial, los monocitos se transforman en macrófagos, los cuales oxidan a las MM-LDL y producen las LDL oxidadas. Este proceso se ve favorecido por la angiotensina II y por la glucosidación previa de las LDL. Los macrófagos captarán a estas LDL oxidadas, proceso mediado por el factor estimulante de colonias de macrófagos (MCSF) y estimulado por la angiotensina II. Los macrófagos así activados pueden estimular la expresión celular de la Enzima Convertidora de Angiotensina (ECA) y la síntesis de angiotensina II, lo que conlleva a un ciclo de retroalimentación positiva. Además, debido a que no existe ningún mecanismo de saturación en los macrófagos, seguirán captando lípidos y se someterán a una sobrecarga que producirá una degeneración en ellos, hasta convertirse en las denominadas células espumosas que finalmente morirán y liberarán los lípidos que formarán el núcleo lipídico, junto con sustancias tóxicas, como enzimas, radicales libres y aniones superóxido. Los productos tóxicos lesionan el endotelio, que en algunas zonas puede ser incluso destruido y desaparecer. Los macrófagos y algunas plaquetas activadas, segregan factores de crecimiento, como el Factor de Crecimiento Derivado de Plaquetas (PDGF, del inglés, Platelet-Derived Growth Factor), que estimulan la proliferación y migración de las células musculares lisas de la túnica media. Esta fase proliferativa aumenta con el descenso de la molécula antiproliferativa Óxido Nítrico (ON) y con el incremento de la angiotensina II. Las células del músculo liso también secretan factores de crecimiento y, además, cubren el núcleo ateromatoso y producen proteínas de matriz (colágeno, elastina y proteoglicanos), que formarán la cubierta fibrosa.42 Una vez formadas, las placas ateroscleróticas pueden crecer lentamente si se mantiene el proceso aterogénico o complicarse de forma repentina (ver Figura 3).53,54,55
El buen funcionamiento endotelial depende de una gran variedad de factores, tales como, la integridad anatómica de las células endoteliales, la correcta señalización entre ellas y una adecuada producción de sustancias vasoactivas. Los factores de riesgo tradicionales como hipercolesterolemia afectan la producción o acción de sustancias vasoactivas claves, como son, el ON endotelial, lo cual, puede desencadenar efectos proaterogénicos y protrombóticos.56,57,58
Se ha observado que principalmente las LDL oxidadas, y en menor medida las LDL no oxidadas, disminuyen la expresión de la Sintasa de Óxido Nítrico endotelial (SONe) que coincide, además, con una menor actividad de la misma.59 Lo anterior favorece el estrés oxidante y la presencia de un estado fuertemente pro-inflamatorio, los cuales, pueden desembocar en alteraciones profundas a nivel vascular.60,61
Existe evidencia de que las arterias de animales ateroscleróticos y de pacientes con hipercolesterolemia o ateroesclerosis producen menores cantidades de ON y presentan una respuesta reducida al estímulo de vasodilatadores dependientes del endotelio. Lo anterior sugiere que aún antes de que se presenten las alteraciones morfológicas típicas de la ateroesclerosis, existe ya una alteración funcional del endotelio como consecuencia del estado pro-inflamatorio.62
Se ha demostrado que elevadas concentraciones de colesterol interrumpen y alteran la estructura y función vascular, ya que se adhiere al revestimiento de la pared vascular, constituido por el endotelio, y puede interferir con la función endotelial, lo cual, conduce a las lesiones, las placas, la oclusión, y la embolia, junto con una reducción en la recuperación y manejo adecuado de la lesión.63,64,65
Es evidente que una inadecuada alimentación se encuentra íntimamente ligada a la prevalencia de los trastornos cardiovasculares. La principal fuente externa del colesterol son los alimentos de origen animal como la carne, las grasas, el huevo y la leche. El exceso en la ingesta de colesterol causa que su concentración aumente en el suero de la sangre y que se acumule en el cuerpo favoreciendo la ateroesclerosis. Aunado a este importante factor, la obesidad, hipertensión, Diabetes mellitus (DM2), tabaquismo, una vida sedentaria e incluso factores socio-económicos, incrementan el riesgo de padecer problemas cardiovasculares a edades tempranas.
Por otro lado, no siempre el consumo de alimentos ricos en grasas e inadecuados estilos de vida son los responsables de altos niveles séricos de colesterol, por lo tanto, el tratamiento eficaz de intervención puede ser problemático cuando se presentan condiciones genéticas, tal es el caso de la Hipercolesterolemia Familiar, la cual, es un ejemplo paradigmático de ateroesclerosis precoz in vivo dependiente del C-LDL.66
Hipercolesterolemia
La dislipidemia más frecuente e importante por su trascendencia etiopatogénica en las enfermedades cardiovasculares, es la hipercolesterolemia, esta condición se define como la presencia de niveles excesivamente elevados de colesterol en sangre. La hipercolesterolemia se caracteriza clínicamente por niveles séricos elevados de colesterol (>240 mg/dL) y elevación de LDL (>190 mg/dL) (ver Tabla 1).65,67
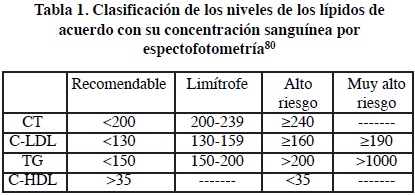
Existen distintas clasificaciones para las hiperlipidemias con diversas variaciones y aunque no existe ninguna completamente satisfactoria, hemos elegido para esta revisión la más utilizada: De acuerdo a criterios etiopatogénicos las hipercolesterolemias se clasifican en:
− Primarias: Base genética o familiar causante de los trastornos lipoproteicos.
− Secundarias: La base de la alteración lipoproteica, esta asociada a una enfermedad adyacente.
Hipercolesterolemias primarias
La Hipercolesterolemia Autosómica Dominante (ADH del inglés: Autosomal Dominant Hypercholesterolemia) quereferimos comúnmente como Hipercolesterolemia Familiar (HF), se caracteriza clínicamente por una elevación constitutiva, de los niveles plasmáticos de C-LDL.68
La HF es un desorden genético con un patrón de herencia autosómico dominante, con una alta prevalencia en la población general, 1 en 500 son portadores heterocigotos y 1 en 1 millón son homocigotos, aunque esta prevalencia puede variar en diferentes poblaciones.69
La HF es causada principalmente por mutaciones en el gen receptor de LDL (rLDL), y menos frecuentemente por mutaciones en los genes para la apo B y la de más reciente identificación PCSK9.70 Esta condición da lugar a un aumento en los niveles plasmáticos de C-LDL. Hasta la fecha, más de 800 mutaciones funcionales diferentes del gen del rLDL se han descrito en todo el mundo. La esperanza de vida se acorta de 20 a 30 años en pacientes con HF y la muerte súbita e infarto de miocardio son las principales causas de muerte.71
La HF homocigota, tiene como génesis, mutaciones en ambos alelos del loci del rLDL. Los pacientes con esta condición, presentan marcadamente niveles elevados de colesterol sérico total (>500 mg/dL, 13 mmol/L) y niveles de colesterol LDL(C-LDL >450 mg/dL, 11.7 mmol/L). El depósito de colesterol insoluble causa xantomas en los tendones de las manos y los pies, y arco corneal en la vida temprana. Aun más relevante, es la aparición de ateromas en la raíz aórtica y válvula, lo que puede conducir a Infarto Agudo de Miocardio (IAM) y muerte súbita antes de la edad de 30 años. La Enfermedad Arterial Coronaria (CAD, del inglés, Coronary Artery Disease) es la más común y más amplia en los pacientes con receptores negativos (mutaciones que eliminan por completo las funciones del receptor) que en aquellos con el tipo de receptor defectuoso(mutaciones que inactivan parcialmente la función del receptor), donde hay receptores residuales activos.72 Como se mencionó anteriormente, la prevalencia de HF homocigota es relativamente baja, sin embargo, algunos grupos étnicos tienen altas prevalencias debido a fenómenos migratorios que resultan en poblaciones genéticamente aisladas. Ejemplos son los francocanadienses residentes en Quebec, los libaneses, los judíos Ashkenazi y algunas comunidades de Sudáfrica. En ellos, la mayor prevalencia es debida a un efector fundador, es decir, los primeros habitantes que colonizaron estas regiones tenían la enfermedad y la transmitieron a las siguientes generaciones. El aislamiento relativo de las poblaciones resulta en un número mayor de casos con formas homocigotas de la enfermedad.73
La HF heterocigota, se presenta en pacientes con una única mutación en el alelo del locis del rLDL, suelen tener un menornivel de colesterol sérico (250-450 mg/dL o 6.5 a 11.6 mmol/L) y C-LDL (200-400 mg/dL o 5.2 a 10.4 mmol/L). Se desarrollan las características clínicas de la HF homocigota, con una tasa menos acelerada, de no existir un tratamiento oportuno, los pacientes pueden sufrir un infarto de miocardio grave y la muerte súbita o con frecuencia otros eventos cardiovasculares en la cuarta o quinta década de la vida. Debido a varios factores hormonales, aproximadamente el 80% de los hombres heterocigotos afectados por esta enfermedad, sufren de enfermedad arterial coronaria, mientras que sólo del 20% al 30% de las mujeres son moderadamente afectadas.72
La HF recesiva tiene características similares al fenotipo clínico de la HF homocigota, pero presentaban un patrón de herencia poco usual (autosómico recesivo). Asimismo la función de los fibroblastos de individuos afectados era casi normal y el fenotipo no correspondía con los locus del rLDL ni de Apo B-100.
Este nuevo padecimiento se designó Hipercolesterolemia Autosómica Recesiva (ARH, por sus siglas en inglés, Autosomal Recessive Hypercholesterolemia). Esta condición es generalmente menos severa (afecta 1 de cada 5 000 individuos) y responde de mejor manera a las terapias convencionales que la HF homocigota. El defecto fisiológico en la HF recesiva es la imposibilidad de algunos tipos de células de mediar la internalización de LDL dependiente del rLDL. Este hallazgo sugería que alteraciones en el tráfico del rLDL (externalización o internalización) pueden causar hipercolesterolemia.74 La disbetalipoproteinemia familiar, es un tipo de hiperlipidemia mixta de carácter autosómico recesivo, en más del 90% una variación en la apo E podría causar la hiperlipidemia. Esta condición se caracteriza por los niveles elevados de colesterol total y triglicéridos (se puede observar ascensos de colesterol de 600 mg/dL y de triglicéridos de 400 a 800 mg/dL, aunque a veces suelen ser mayores), debido a altos niveles plasmáticos de quilomicrones y remanentes de VLDL enriquecidos de ésteres de colesterol y apo E.75 La apo E, es un componente importante de los quilomicrones y VLDL remanentes, sirve como un ligando para la captación mediada por los receptores de estas partículas por el hígado. Las mutaciones apo E conducen a una alteración en la depuración de los remanentes de las lipoproteínas, por los receptores de lipoproteínas hepáticas. Hay tres variantes genéticas comunes de la apo E: Apo E-2 (Arg158→Cis), apo E-3 (Cis112, Arg158) y apo E-4 (Cis112→Arg). Estas isoformas están codificadas por tres alelos codominantes que están situados en un locus del gen del cromosoma 19. La manifestación clínica es la formación prematura de placa de ateroma e infarto de miocardio, accidente cerebrovascular o enfermedad arterial periférica.76
La Hipercolesterolemia Poligénica (HP) es la forma más común de hipercolesterolemia primaria y se atribuye a la intervención de distintos genes, el defecto de estos genes repercute en un aumento en las concentraciones de C-LDL. Está condición estrechamente modulada por factores ambientales y los rasgos hereditarios son menos evidentes. Con frecuencia, las personas con HP tienen historia familiar de enfermedad coronaria prematura y antecedentes familiares de hipercolesterolemia. Las personas jóvenes con HP pueden tener las concentraciones de colesterol normales o ligeramente elevadas y la hipercolesterolemia se puede expresar más tardíamente. El diagnóstico debe sospecharse en cualquier persona con cifras de colesterolemia de 280 a 320 mg/dL y con concentraciones de triglicéridos normales.77
La Hiperlipidemia Familiar Combinada (HLFC) es una dislipidemia común que conlleva un alto riesgo de enfermedad cardiovascular. La prevalencia de HLFC en la población general se ha informado aproximadamente de hasta 6%, mientras que en las familias con tasas prematuras de enfermedades cardíacas coronarias tienen una prevalencia de 15 a 20%.78 De hecho, se estima que aproximadamente una de cada cinco pacientes que sobrevivieron un infarto de miocardio prematuro es un portador HLFC. Aunque la transmisión sigue un patrón hereditario autosómico dominante, un único gen o conjunto de genes no ha sido identificada para la HLFC.
Los estudios metabólicos indican que el defecto principal en HLFC es el aumento en la tasa de producción de lipoproteínas ricas en triglicéridos, principalmente las VLDL derivadas del hígado, con la consiguiente hiperlipidemia caracterizada por incrementos plasmáticos de triglicéridos y colesterol.79 La HLFC comparte algunas características con el síndrome metabólico, ambas entidades pueden tener niveles elevados de triglicéridos, con LDL pequeñas y densas, sin embargo, típicamente en la HLFC se encuentran concentraciones altas de apo B. Se ha reportado que el patrón cambiante de esta dislipidemia (hipertriacilgliceridemia con hipoalfalipopro-teinemia o aumento de C-LDL guarda una estrecha relación con la cantidad total de grasa visceral y de resistencia a la insulina. Para establecer el diagnóstico con certidumbre se requiere el estudio del mayor número posible de miembros de la familia. La ausencia de xantomas es un requisito indispensable para considerar un caso como afectado, pero deben tomarse otras consideraciones para el diagnóstico, por ejemplo, la concentración de la apo B-100 generalmente se encuentra por encima del percentil 90 para el grupo étnico correspondiente. La elevación de C-LDL y/o de los triglicéridos es moderada (pocas veces supera 300 mg/dL), sin embargo, al combinarse con otras causas de dislipidemia pueden observarse niveles extremadamente altos de colesterol y/o triglicéridos.77 La estrategia farmacológica sigue las siguientes condiciones: Si predomina la elevación del CT sobre los TG, el tratamiento de elección son las estatinas. Si predominan los TG (cifras >500 mg/dL) sobre el CT el tratamiento son los fibratos. En ocasiones es necesario el tratamiento combinado, dependiendo del riesgo cardiovascular del paciente.80
La hipercolesterolemia tipo B (Apo B Defectuosa Familiar, FDB, del inglés, Familiar Defective Apolipoprotein B) es otro defecto genético que altera el transporte de colesterol, es una enfermedad autosómica codominante, debida a mutaciones en el gen de la apo B-100.
La mayoría de las mutaciones en apo B-100, se localizan en una región del exón 26 que flanquea con el codón 3 500 del gen. Debido a que esta región codifica para el dominio de unión de Apo B-100 al receptor de LDL, las mutaciones afectan el transporte de LDL al interior de la célula, conduciendo al incremento de la concentración de LDL circulante. La primera mutación descrita en el gen apo B-100 y la más frecuente en la mayoría de poblaciones estudiadas es una transición G→A que resulta en una sustitución Arg3 500→Gln en el dominio de unión de apo B-100 al receptor de C-LDL. Otras mutaciones descritas para hipercolesterolemia tipo B son sustituciones Arg3 500→Trp (CGG→TGG), el mismo codón señalado anteriormente 21-23 y Arg3 531→Cis, ambas afectando el dominio de unión de apo B-100 al rLDL. Las mutaciones en apoB-100 también se asocian a elevado riesgo de enfermedad cerebro vascular y los niveles de colesterol están en el mismo rango de lo encontrado en la HF, aunque pueden ser ligeramente menores.81 Los pacientes presentan cuadros clínicos menos severos con una aterogenicidad menor que los observados en pacientes con mutaciones en rLDL. La prevalencia de la FDB es bastante más baja que la debida a defectos en rLDL, aunque varía mucho (2-12%) de acuerdo a la población estudiada. La prevalencia más alta se encuentra en Europa Central y Suiza (1:200 adultos), mientras que en otros países como Finlandia, no ha sido detectada.68
La proteína convertasa PCSK9 juega un papel clave en lahomeostasis del colesterol mediante la unión al rLDL. El gen PCSK9 fue identificado en el cromosoma 1-proproteína subtilisina convertasa /Kexin tipo IX. Este gen codifica para una proteína de 694 aminoácidos que pertenecen a una familia de proproteína subtilisina convertasa.82 La PCSK9 es secretada por los hepatocitos y parece regular la degradación intracelular del rLDL. Las mutaciones con ganancia de función son capaces de disminuir su densidad en la superficie celular, afectando la depuración plasmática de C-LDL y generando un fenotipo semejante a la HF. Actualmente la estrategia experimentar para contrarestar el efecto deletéreo de esta mutación, radica en las mutaciones con pérdida de función para PCSK9 ya que se reduciría constitutivamente el nivel de C-LDL plasmático y el riesgo de enfermedad coronaria por lo que ha generado gran interés la posibilidad de regular farmacológicamente su función.83
El término "dislipemias secundarias" se utiliza para describir las alteraciones cuantitativas y/o cualitativas en el metabolismo de las lipoproteínas asociadas a una enfermedad adyacente. Constituyen la mayoría de los casos de dislipidemia en adultos. La causa más frecuente es el estilo de vida sedentario con ingesta elevada de grasas saturadas y colesterol; otras causas son la DM2, el consumo excesivo de alcohol, la insuficiencia renal crónica, el hipotiroidismo, la cirrosis hepática primaria y algunos fármacos como las tiacidas, retinoides, antirretrovirales, estrógenos, progestágenos y glucocorticoides.84 Al igual que las dislipidemias primarias, las dislipidemias secundarias involucran cambios en el metabolismo lipídico, lo que significativamente aumenta el riesgo de eventos cardiovasculares, así como para otras complicaciones específicas de la enfermedad. Por ejemplo, la dislipidemia asociada a enfermedades renales crónicas puede acelerar el deterioro de la función renal en pacientes con insuficiencia renal, mientras que la dislipidemia diabética puede aumentar el riesgo de pancreatitis aguda en individuos genéticamente predispuestos.
Por otra parte, puesto que la mejora de la enfermedad subyacente (sí es posible) da lugar generalmente a la corrección (al menos en parte) de anormalidades en los lípidos, la correcta identificación de una causa secundaria de la dislipidemia puede ayudar a evitar el tratamiento hipolipemiante innecesario.80,85
Factores dietéticos y farmacológicos
-Modificación de estilo de vida
En los pacientes con HF, deben implementarse modificaciones en los estilo de vida para reducir el C-LDL y factores de riesgo de enfermedad cerebro vascular, a pesar de que el grado de reducción de C-LDL es modesto y variable siendo aproximadamente de 10%. Una dieta que contiene menos de 7% de grasa saturada y menos de 200 mg de colesterol debe ser aconsejado. Aunque existen distintos puntos de vista, hay consenso en que deben consumirse preferentemente frutas y vegetales frescos, que son ricos en nutrientes como vitaminas y minerales, y abundantes en fibra dietética que comprende la parte de los carbohidratos que no se absorben y, por tanto, aportan pocas calorías.
Los pacientes deberían ser alentados a lograr y mantener un peso saludable a través de la actividad física y una adecuada ingesta de calorías. El consumo de alcohol y el tabaquismo debe ser restringido ya que se asocian con las enfermedades cardiovasculares en pacientes con hipercolesterolemia.86,87
Fitoesteroles
Los fitoesteroles y sus formas reducidas, los fitoestanoles, son esteroles de origen vegetal ampliamente distribuidos en la naturaleza y cuya estructura es muy similar a la del colesterol (ver Figura 4).

Desde hace años se conoce que estos esteroles producen efectos hipocolesterolémicos cuando son ingeridos en el rango de 1-3 g/día, por lo cual se les considera como importantes aliados en la prevención de las enfermedades cardiovasculares, siendo su consumo indicado para individuos con hipercolesterolemias leves o moderadas. El efecto hipocolesterolémico de los fitoesteroles y de los fitoestanoles es atribuido a tres acciones metabólicas: a) Inhibición de la absorción intestinal de colesterol por competencia en la incorporación del colesterol a las micelas mixtas,88 b) disminución de la esterificación del colesterol en los enterocitos al inhibir la actividad de la enzima ACAT2 y, c) estimulación del eflujo de colesterol desde los enterocitos hacia el lumen intestinal al aumentar la actividad y la expresión de un transportador de tipo ABC. La acción conjunta de los esteroles y/o estanoles sobre estos mecanismos produce una disminución del colesterol total plasmático y del C-LDL, sin modificar los niveles del C-HDL. Por su efecto reductor del colesterol, estos compuestos se han incorporado a margarinas y productos lácteos y se han introducido en la cadena alimentaria como alimentos funcionales para el tratamiento de la hipercolesterolemia moderada.27,89
Probióticos
La flora intestinal está directamente relacionada con la salud del hospedero. Actualmente es posible manipular la flora intestinal para asegurar efectos benéficos sobre la salud humana. Esta manipulación se logra gracias a los probióticos, cuyas bacterias vivas incorporadas a ciertos alimentos, son capaces de alcanzar el intestino delgado y colon. De esta manera se incrementa tanto la densidad bacteriana y la actividad metabólica de la flora intestinal. Dentro de los efectos benéficos, se ha logrado disminuir la intolerancia a la lactosa, prevenir o disminuir diarreas infecciosas, prevenir el desarrollo de cáncer de colon y desarrollar un efecto inmunoprotector. En el mismo sentido, se sabe que diversas bacterias de la flora intestinal (Lactobacillus acidophilus ATCC 43121, Lactobacillus plantarum 9-41-A, Lactobacillus fermentum M1-16, Lactobacillus casei ASCC 292) poseen un efecto hipocolesterolemiante.90,91,92
Desde que Mann y Spoerry93 descubrieron los efectos hipocolesterolémicos de la leche fermentada por nativos de la tribu Massai, la relación entre las bacterias ácido lácticas y el colesterol sérico se ha convertido en un foco de gran interés. El mecanismo de acción hipocolesterolemiante no está totalmente establecido, sin embargo, se han propuesto varias hipótesis para explicar estos resultados: (1) Las bacterias intestinales metabolizan colesterol por lo que reduce la cantidad de colesterol disponible para la absorción, (2) parte del colesterol puede ligarse a la superficie bacteriana celular o incorporarse en las membranas celulares bacterianas o convertido en coprostanol por la colesterol reductasa, que es producida por cepas de lactobacilos, (3) la inhibición de la formación de micelas por ciertas cepas probióticas, (4) los ácidos grasos de cadena corta producidos durante la fermentación selectiva de los alimentos por la microflora bacteriana intestinal, puede disminuir los niveles de colesterol en plasma por la competición de micelas, y (5) algunas especies de bacterias tienen la capacidad de desconjugar las sales biliares, obligando al organismo a producir más sales biliares y por ende, se incrementa el consumo de colesterol endógeno, el cual es la materia prima de las sales biliares. Estas alternativas se consideran para pacientes con reacciones alérgicas a los compuestos de medicamentos hipolipemiantes, aunado a que evitan los efectos adversos que estos pudieran tener.93,94
La hipercolesterolemia y terapias farmacológicas
Actualmente los fármacos de primera elección para el tratamiento de pacientes en que predomina el incremento plasmático de colesterol son las estatinas.
Las estatinas son inhibidores de la 3-hidroxi-3-metilglutaril coenzima A reductasa (HMG-CoAR), mejor conocidos como estatinas que actúan mediante la interferencia de la síntesis del ácido mevalónico a partir de la HMG-CoAR, lo que limita la síntesis de colesterol.
Las estatinas son poderosos fármaco hipolipemiantes, que a la vez son generalmente bien toleradas. Actúan inhibiendo parcial y temporalmente a la reductasa de la 3-hidroxi-3-metilglutaril coenzima A y con ello disminuyen la síntesis de novo del colesterol en el hepatocito.96 De las estatinas disponibles en México, la de mayor uso y efectividad es la pravastatina, ampliamente estudiada, que en grandes ensayos clínicoscontrolados ha demostrado su efectividad para disminuir los niveles séricos de colesterol, así como la disminución de nuevos eventos de síndrome coronario agudo y muerte secundaria debida a cardiopatía isquémica. Además, reduce la placa ateromatosa, disminuye hasta 16% el colesterol total y 22% las LDL después de ocho semanas de tratamiento con 10 mg/día.
La pravastatina es segura y efectiva, absorbiéndose rápidamente y alcanzando niveles plasmáticos máximos a las 1 a 1.5 horas posterior a su ingestión, se metaboliza en hígado y se excreta por orina (20%) y heces (70%). Está contraindicada en falla hepática y reacciones adversas, así como en embarazo y lactancia. La dosis usual es de 10 o 20 mg al día.97
Las resinas, la niacina, fibratos, entre otros, han sido los fármacos que clásicamente se han asociado con las estatinas para incrementar su eficacia sobre los valores del colesterol. No obstante, suelen presentar efectos secundarios con frecuencia, lo que dificulta que se alcancen las dosis necesarias para la obtención de los objetivos terapéuticos en pacientes de alto riesgo coronario.98 En la Tabla 2 se presenta el mecanismo de acción, efectos sobre los lípidos y efectos adversos, de las terapias actualmente utilizadas para la disminución de los niveles de colesterol plasmático.
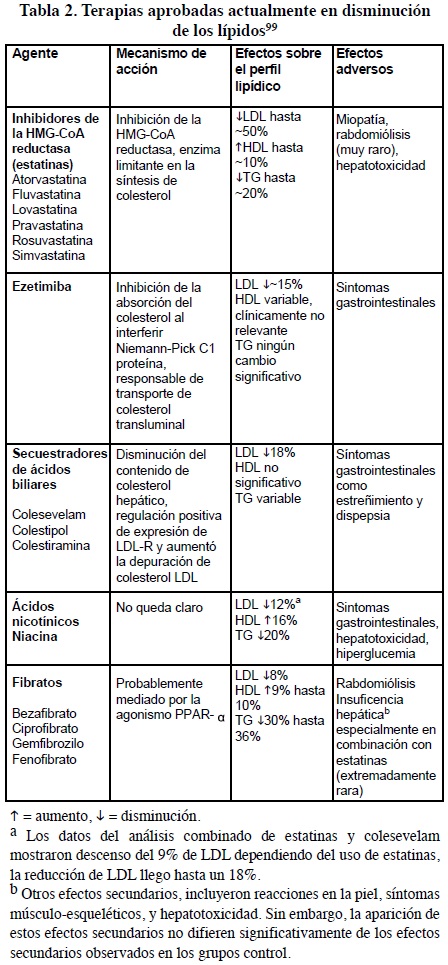
HDL lipoproteínas de alta densidad, HMG-CoA 3-hidroxi-3metilglutaril coenzima A, LDL lipoproteínas de baja densidad; LDLR receptor de lipoproteína de baja densidad, PPAR-α peroxisoma proliferador activado del receptor-α, TG triglicéridos.
La ezetimiba es un fármaco que se indica generalmente en la hipercolesterolemia primaria. A través de su prescripción y su consumo se reducen los niveles de CT y de LDL. La ezetimiba es un potente inhibidor de la absorción del colesterol, destacable por su eficacia, seguridad y grado de desarrollo clínico.30 Los inhibidores de la absorción del colesterol a diferencia de los fitoesteroles, que deben ingerirse en dosis altas (gramos), la ezetimiba es activa (y más eficaz) en dosis muy pequeñas (miligramos). También difiere de otros agentes hipolipemiantes que actúan en el intestino, como las resinas de intercambio aniónico, que estimulan el catabolismo del colesterol al fijar ácidos biliares, esto por interacción física, lo que requiere dosis de gramos (hasta 24 g al día en el caso del Questran: Resina de colestiramina) para impedir su reabsorción.8,9,100
La ezetimiba actúa inhibiendo el transporte activo del colesterol mediado por el receptor Niemann-Pick C1. Este mecanismo disminuye el aporte de colesterol al hígado, aumentando la sobreexpresión de receptores hepáticos de LDL y conduce a una reducción del C-LDL de aproximadamente 18-20%, con mínimos cambios sobre C-HDL (aumento 1–3%), una reducción de los TG menor del 10% y sin ningún efecto secundario grave.101
Terapia combinada
Como se mencionó anteriormente, la ezetimiba reduce la llegada de colesterol al hígado determinando un aumento compensatorio de la síntesis y las estatinas inhiben marcadamente la síntesis, oponiéndose al marcado efecto contrarregulador hepático.
Por lo tanto, la administración conjunta de ambos fármacos tiene efectos sinérgicos en la reducción de la colesterolemia. La coadministración de la ezetimiba y estatinas es una nueva modalidad de tratamiento hipocolesterolemiante altamente eficaz y seguro, que permite alcanzar los objetivos de C-LDL en una proporción mucho mayor de pacientes de alto riesgo coronarios.102,103
Los resultados de cuatro estudios en fase III en los que se compararon los efectos del tratamiento con una estatina administrada a dosis crecientes hasta la máxima recomendada (lovastatina y pravastatina: 10, 20 o 40 mg; simvastatina y atorvastatina: 10, 20, 40 y 80 mg) y los de la administración conjunta de ezetimiba, 10 mg/día (añadida a cada una de las dosis de cada estatina) durante 12 semanas han precisado claramente que la eficacia de la coadministración de ezetimiba con cualquier dosis de estatina es superior a la de la estatina de manera individual. En estos cuatro estudios se incluyeron a un total de 2 382 pacientes con hipercolesterolemia primaria (C-LDL entre 145 y 250 mg/dL), y en todos ellos la mayor eficacia de la coadministración se observó con todas las dosis de estatina. Además, la administración concomitante de 10 mg de ezetimiba al día con la dosis más baja de cada estatina sería tan eficaz o más en la reducción del C-LDL que la monoterapia con estatinas a la dosis más alta.104,105,106
Finalmente, es importante recalcar que, todas las estrategias para mantener los niveles de colesterol en valores recomendables, tanto las no farmacológicas que incluyen cambios en los hábitos alimenticios y en el estilo de vida, y las farmacológicas, mejorarían la calidad de vida de los pacientes con problemas de colesterol; los fármacos no curan la hipercolesterolemia, sólo contribuyen a mejorar los resultados dentro de un tratamiento integral, disminuyendo la síntesis endógena o bloqueando la absorción de grasa, la farmacoterapia bajo ninguna circunstancia sustituirá al tratamiento integral con modificación de la dieta y el estilo de vida.
Conclusiones
El colesterol forma parte de las membranas celulares y es precursor de hormonas esteroideas, ácidos biliares y de la formación de la vitamina D. A pesar de su importancia biológica, es evidente que elevadas concentraciones plasmáticas de colesterol ocasionan alteraciones profundas, tales como, la hipercolesterolemia, la cual promueve, la disfunción endotelial causada por la ateroesclerosis. La evolución de la ateroesclerosis representa el principal factor de riesgo para el desarrollo de enfermedades cardiovasculares.
La hipercolesterolemia en general, es un problema grave de Salud Pública que requiere de estudios más detallados en cuanto a la homeostasia del colesterol, ya que la implementación de nuevas estrategias dietéticas, farmacológicas o cambios en el estilo de vida, permitirán mantener en niveles recomendables el colesterol plasmático.
La administración conjunta de ezetimiba y estatinas es una nueva alternativa eficaz y segura que permite reducir significativamente los niveles de C-LDL, que cuando se administran por separado. El efecto terapéutico de estos dos fármacos logra disminuir en gran medida la incidencia de las enfermedades cardiovasculares; finalmente, todas las estrategias para mantener los niveles de colesterol en valores recomendables, tanto las no farmacológicas que incluyen cambios en los hábitos alimenticios y en el estilo de vida, y las farmacológicas, mejorarían la calidad de vida de los pacientes con problemas de colesterol, finalmente es importante recalcar que la farmacoterapia no sustituye al tratamiento integral con modificación de la dieta y el estilo de vida.
Agradecimientos
Al Consejo Nacional de Ciencia y Tecnología (CONACyT) por la beca otorgada a Octavio Maldonado Saavedra (CVU 273217), estudiante del Centro de Investigaciones Biomédicas-Doctorado en Ciencias Biomédicas-UV.
Referencias
1. Pasqualini JR. Enzymes involved in the formation and transformation of steroid hormones in the fetal and placental compartments. J Steroid Biochem Mol Biol. 2005;97:401-415. [ Links ]
2. Navarro V, Zabala A, Gómez S, Portillo M. Metabolismo del colesterol: bases actualizadas. Rev Esp Ob. 2009; 7(6): 360-384. [ Links ]
3. Horton JD, Goldstein JL, Brown MS. SREBPs: activators of the complete program of cholesterol and fatty acid synthesis in the liver. J Clin Invest. 2002; 109:1125-31. [ Links ]
4. Vance DE, Van den Bosch H. Cholesterol in the year 2000. Biochem Biophys Acta. 2000; 15(1529):1-8. [ Links ]
5. Yuan G, Wang J, Hegele RA. Heterozygous familial hypercholesterolemia: an underrecognized cause of early cardiovascular disease. Can Med Assoc J. 2006;174:1124-1129. [ Links ]
6. Ascaso JF. Avances en el tratamiento de la hipercolesterolemia. Endocrinol Nutr. 2010; 57(5):210-219. [ Links ]
7. Magnussen CG, Raitakari OT, Thomson R, Juonala M, Patel DA, Viikari JS, Marniemi J, Srinivasan SR, Berenson GS, Dwyer T, Venn A. Utility of currently recommended pediatric dyslipidemia classifications in predicting dyslipidemia in adult-hood: Evidence from the Childhood Determinants of Adult Health (CDAH) study, Cardiovascular Risk in Young Finns Study, and Bogalusa Heart Study. Circulation. 2008; 117:32-42. [ Links ]
8. Lee MH, Lu K, Hazard S, Yu H, Shulenin S, Hidaka H, Lee MH, Lu K, Hazard S, Yu H, Shulenin S, Hidaka H, Kojima H, Allikmets R, Sakuma N, Pegoraro R, Srivastava AK, Salen G, Dean M, Patel SB. Identification of a gene, ABCG5, important in the regulation of dietary cholesterol absorption. Nat Genet 2001; 27(1):79-83. [ Links ]
9. Ballantyne C. Ezetimibe: efficacy and safety in clinical trials. Eur Heart J. 2002; 4(J):J5-15. [ Links ]
10. Brodsky JL, Fisher EA. The many intersecting pathways underlying apolipoprotein B secretion and degradation. Trends Endocrinol Metab. 2008; 19(7):254-9. [ Links ]
11. Jonas A. Lecithin cholesterol acyltransferase. Biochem Biophys Acta. 2000; 1529: 245-256. [ Links ]
12. Bloch K. The Biological Synthesis of Cholesterol. Science. 1965; 150:19-28. [ Links ]
13. Atshaves BP, McIntosh AL, Martin GG, Landrock D, Payne HR, Bhuvanendran S, Landrock KK, Lyuksyutova OI, Johnson JD, Macfarlane RD, Kier AB, Schroeder F. Overexpression of sterol carrier protein-2 differentially alters hepatic cholesterol accumulation in cholesterol-fed mice. J Lipid Res. 2009; 50(7); 1429-47. [ Links ]
14. Thompson A, Di Angelantonio E, Sarwar N, Erqou S, Saleheen D, Dullaart R. P, Keavney B,Ye Z, Danesh J.Association of cholesteryl ester transfer protein genotypes with CETP mass and activity, lipid levels, and coronary risk. Jama 2008; 299(23): 2777-88. [ Links ]
15. Turley SD. The role of Niemann-Pick C1–Like 1 (NPC1L1) in intestinal sterol absorption. J Clin Lipidol. 2008; 2(2):S20-S28. [ Links ]
16. Li T, Chiang JY. Regulation of Bile Acid and Cholesterol Metabolism by PPARs. PPAR Res. 2009; 2009:501739. [ Links ]
17. Méndez N, Uribe M. Inhibición de la absorción intestinal de colesterol. Una nueva estrategia para el tratamiento médico de la litiasis biliar de colesterol. Med Univer. 2008;10(41):230-4. [ Links ]
18. Turley SD, Dietschy JM. Sterol absorption by the small intestine. Curr Opin Lipidol. 2003; 14:233-40. [ Links ]
19. Schmitz G, Langmann T, Helmerl S. Role of ABCG1 and other ABCG family members in lipid metabolism. J Lipid Res. 2001; 42:1513-20. [ Links ]
20. Betters J, Yu L. NPC1L1 and Cholesterol Transport. FEBS Lett. 2010; 584(13):2740–2747. [ Links ]
21. Altmann SW, Davis HR Jr, Zhu LJ, Yao X, Hoos LM, Tetzloff G, Lyer SP, Maguire M, Golovko A, Zeng M, Wang L, Murgolo N,Graziano MP. Niemann-Pick C1 Like 1 protein is critical for intestinal cholesterol absorption. Science. 2004; 303:1201-4. [ Links ]
22. Garcés C. Regulación de la absorción intestinal de colesterol: el papel protagonista de NPC1L1 y suspolimorfismos funcionales. Clin Invest Arterioscl. 2008; 20:(5)207-9. [ Links ]
23. Jakulj L, Trip MD, Sudhop T, Von Bergmann K, Kastelein JJ, Vissers MN. Inhibition of cholesterol absorption by the combination of dietary plant sterols and ezetimibe: effects on plasma lipid levels. J Lipid Res. 2005; 46(12):2692-8. [ Links ]
24. Jakulj L, Vissers MN, Tanck MW, Hutten BA, Stellaard F, Kastelein JJ, Dallinga-Thie GM. ABCG5/G8 polymorphisms and markers of cholesterol metabolism: systematic review and meta-analysis. J Lipid Res. 2010; 51(10): 3016-3023. [ Links ]
25. Garcia-Rios A, Perez-Martinez P, Fuentes F, Mata P, Lopez-Miranda J, Alonso R, Rodriguez F, Garcia-Olid A, Ruano J, Ordovas JM, Perez-Jimenez F. Genetic Variations at ABCG5/G8 Genes Modulate Plasma Lipids Concentrations in Patients with Familial Hypercholesterolemia. Atherosclerosis. 2010; 210(2):486-492. [ Links ]
26. Chao H, Zhou M, McIntosh A, Schroeder F, Kier A. ACBP and cholesterol differentially alter fatty acyl CoA utilization by microsomal ACAT. J Lipid Res. 2003; 44(1):72-83. [ Links ]
27. Valenzuela B, Ronco M. Fitoesteroles y fitoestanoles: aliados naturales para la protección de la salud cardiovascular. Rev Chil Nutr. 2004; 21(1):161-169. [ Links ]
28. Wetterau JR, Aggerbeck LP, Bouma M-E, Eisenberg C, Munck A, Hermier M, Schmitz J, Gay G, Rader DJ, Gregg RE. Absence of microsomal triglyceride transfer protein in individuals with abetalipoproteinemia. Science. 1992;258:999-1001. [ Links ]
29. Cohn J, Kamili A, Wat E, Chung R, Tandy S. Dietary Phospholipids and Intestinal Cholesterol Absorption Nutrients. 2010; 2 (2):116-127. [ Links ]
30. Ros E. Doble inhibición del colesterol: papel de la regulación intestinal y hepática. Rev Esp Cardiol. 2006; 6:52-62. [ Links ]
31. Janowski BA. The hypocholesterolemic agent LY295427 upregulates INSIG-1, identifying the INSIG-1 protein as a mediator of cholesterol homeostasis through SREBP. Proc Natl Acad Sci USA. 2002; 99:12675-12680. [ Links ]
32. Zanlungo S, Rigotti A, Nervi F. Hepatic cholesterol transport from plasma into bile: implications for gallstone disease. Curr Opin Lipidol. 2004;15: 279-286. [ Links ]
33. Nelson DL, Cox MM. Lehininger Principios de Bioquímica. 4ª. Edición. Editorial Omega; 2005, p. 816-829. [ Links ]
34. Campbell MK, Farrel SO. Bioquímica. 4ª. Edición. Editorial Thomson; 2004, p. 601-613. [ Links ]
35. Lin Y, Mousa S, Elshourbagy N, Mousa S. Current status and future directions in lipid management: emphasizing low-density lipoproteins, high-density lipoproteins, and triglycerides as targets for therapy. Vasc Health Risk Manag. 2010; 6:73–85. [ Links ]
36. Tomkin GH, Owens D. The Chylomicron: Relationship to Atherosclerosis Gerald. Int J Vasc Med. 2012; 2012:784536. [ Links ]
37. Armengol S, Botham K, Lawson Ch. The Oxidative State of Chylomicron Remnants Influences Their Modulation of Human Monocyte Activation. Int J Vasc Med. 2012. doi:10.1155/2012/942512. [ Links ]
38. Napolitano M, Kruth HS, Bravo E. Phospholipase A2 Mediates Apolipoprotein-Independent Uptake of Chylomicron Remnant-Like Particles by Human Macrophages. Int J Vasc Med. 2012; 2012: 501954. [ Links ]
39. Contreras-Leal É, Santiago-García J. Obesidad, síndrome metabólico y su impacto en las enfermedades Cardiovasculares. Rev Biomed. 2011; 22:103-115. [ Links ]
40. Córdova L. Consecuencias de la reabsorción enteral de lípidos ¿Se justifica el tratamiento combinado estatina-ezetimiba de primera línea?. Arch Cardiol Mex. 2004; 74: 2. [ Links ]
41. Zhang L, Song J, Cavigiolio G, Ishida BY, Zhang S, Kane JP, Weisgraber KH, Oda MN, Rye KA, Pownall HJ, Re G. Morphology and structure of lipoproteins revealed by an optimized negative-staining protocol of electron microscopy. J Lipid Res. 2011; 52(1)175-184. [ Links ]
42. Badimóna J, Ibáñez B. Incremento de las HDL como arma terapéutica en la aterotrombosis. Rev Esp Cardiol. 2010; 63(3):323-33. [ Links ]
43. Singh V, Sharma R, Kumar A, Deedwania P. Low high-density lipoprotein cholesterol: current status and future strategies for management. Vasc Health Risk Manag. 2010; 6:979–996. [ Links ]
44. Fernández V, Morales IM, Molero-Conejo E. Niveles de Apoproteínas B, A1 y CIII como Marcadores de riesgo Cardiovascular en adolescentes delgados y obesos. Invest Clin. 2004; 45(1): p. 29-42. [ Links ]
45. Ryan TM, Teoh CL, Griffin MD, Bailey MF, Schuck P, Howlett GJ. Phospholipids enhance nucleation but not elongation of apolipoprotein C-II amyloid fibrils. J Mol Biol. 2010; 399(5):731–740. [ Links ]
46. Zheng C, Khoo C, Furtado J, Sacks FM. Apolipoprotein C-III and the Metabolic Basis for Hypertriglyceridemia and the Dense Low-Density Lipoprotein Phenotype. Circulation. 2010; 121(15):1722-1734. [ Links ]
47. Nicholls S, Tang W, Scoffone H, Brennan D, Hooman J. Lipoprotein(a) levels and long-term cardiovascular risk in the contemporary era of statin therapy. J Lipid Res. 2010; 51(10):3055-3061. [ Links ]
48. Marcel E. Niveles de lipoproteínas en pacientes con enfermedad cerebrovascular oclusiva aterotrombótica. Rev Mex Patol Clin. 2011; 58(3):156-168. [ Links ]
49. BeLue R, Lanza ST, Figaro MK. Lifestyle therapy changes and hypercholesterolemia: identifying risk groups in a community sample of Blacks and Whites. Ethn Dis. 2009;19 (2):142-7. [ Links ]
50. Tunón J, Egido J. Endothelial dysfunction, inflammation and statins: new evidences. Rev Esp Cardiol. 2004; 57(10):903-905. [ Links ]
51. Pullinger CR, Eng C, Salen G, Shefer S, Batta AK, Erickson SK, Verhagen A, Rivera CR, Mulvihill SJ, Malloy MJ, Kane JP. Human cholesterol 7alpha-hydroxylase (CYP7A1) deficiency has a hypercholesterolemic phenotype. J Clin Invest. 2002; 110(1):109-17. [ Links ]
52. Aguilar-Salinas CA, Olaiz G, Valles V, Ríos JM, Gómez FJ, Rull JA, Rojas R, Franco A, Sepulveda J. High prevalence of low HDL cholesterol concentrations and mixed hyperlipidemia in a Mexican nationwide survey. J Lipid Res. 2001; 42:1298-307. [ Links ]
53. Li Z, Li L, Zielke HR, Cheng L, Xiao R, Crow WG, Stetler-Stevenson J, Froehlich. Increased expression of 72-kd type IV collagenase (MMP-2) in human aortic atherosclerotic lesions. Am J Pathol. 1996;148:121-8. [ Links ]
54. Malpartida F, Vivancos R, Urbano C, Mora J. Inflamación y placa inestable. Arch Cardiol Mex. 2007; 77(S4):16-22. [ Links ]
55. Simionescu M. Implications of early structural-functional changes in the endothelium for vascular disease. Arterioscler Thromb Vasc Biol. 2007; 27:266-274. [ Links ]
56. García C, Borja AM, Badimon J. Colesterol HDL y su papel en la ateroesclerosis: ¿Unde venis, quo vadis?. Rev Fed Arg Cardiol 2008; 37: 94-105. [ Links ]
57. Hanson GK. Atherosclerosis and coronary artery disease. N Engl J Med. 2005; 352:1685-1695. [ Links ]
58. Badimóna L, Vilahurb G, Padró T. Lipoproteínas, plaquetas y aterotrombosis. Rev Esp Cardiol. 2009; 62(10):1161-78. [ Links ]
59. Rizo GO, Ramírez JI, Gómez YC. Enfoque actual sobre la fisiopatología del síndrome coronario agudo. Rev Cub Med. 2009; 48(3):71-87. [ Links ]
60. Cachofeiro R, Vázquez N, Cediel G, Sanz R, Olivares E, Lahera J. Hipercolesterolemia y disfunción endotelial: mecanismos implicados. Hipertensión. 2003; 20(3):116-26. [ Links ]
61. Stokes KY, Calahan L, Russell JM, Gurwara S, Granger DN. Role of platelets in hypercholesterolemia-induced leukocyte recruitment and arteriolar dysfunction. Microcirculation. 2006; 13:377-388. [ Links ]
62. Simón A, Castro A, Kaski JK. Avances en el conocimiento de la disfunción endotelial y su aplicación en la práctica clínica. Rev Cub Med 2001; 40(3):212-22. [ Links ]
63. Buitrago L, Delgadillo A, Guzmán N, Fernández N, Mejía I, González MP, Montes F, Echeverri D. Disfunción endotelial inducida por hipercolesterolemia: estudio in vitro en un modelo animal. Rev Med. 2005; 13(1):37-44. [ Links ]
64. Boodhwani M, Nakai Y, Mieno S, Voisine P, Bianchi C, Araujo EG, Feng J, Michael K, Li J, Sellke FW. Hypercholesterolemia impairs the myocardial angiogenic response in a swine model of chronic ischemia: role of endostatin and oxidative stress. Ann Thorac Surg. 2006; 81:634-641. [ Links ]
65. Stapleton PA, Goodwill AG, James ME, Brock RW, Frisbee JC. Hypercholesterolemia and microvascular dysfunction: interventional strategies. J Inflamm (Lond). 2010; 7:54. [ Links ]
66. Drolet MC, Plante E, Battistini B, Couet J, Arsenault M. Early endothelial dysfunction in cholesterol-fed rabbits: a non-invasive in vivo ultrasound study. Cardiovasc Ultrasound. 2004; 2:10. [ Links ]
67. NORMA Oficial Mexicana NOM-037-SSA2-2002, Para la prevención, tratamiento y control de las dislipidemias. [ Links ]
68. Stoll M, Lorenzo M, Raggio V, Esperón P, Zelarayan M. Previniendo el infarto en el adulto joven: GENYCO, un registro nacional de hipercolesterolemia familiar. Rev Urug Cardiol 2011; 26:16-26. [ Links ]
69. Fahed AC, Nemer GM. Familial Hypercholesterolemia: The Lipids or the Genes?. Nutr Metab (Lond). 2011; 8: 23. [ Links ]
70. Bender R, Bell DA, Hooper AJ, Edwards G, van Bockxmeer FM, Watts GF, Burnett JR. Screening for familial hypercholesterolaemia. Pathology. 2012; 44(2):122-8. [ Links ]
71. Mata N, Alonso R, Badimón L, Padró T, Fuentes F, Muñiz O, Perez-Jiménez F, López-Miranda J, Díaz J, Vidal JI, Barba A, Piedecausa M, Sanchez JF, Irigoyen L, Guallar E, Ordovas JM, Mata P. Clinical characteristics and evaluation of LDL-cholesterol treatment of the Spanish Familial Hypercholesterolemia Longitudinal Cohort Study (SAFEHEART). Lipids Health Dis. 2011; 10:94. [ Links ]
72. Al-Allaf FA, Coutelle C, Waddington SN, David AL, Harbottle R, Themis M. LDLR-Gene therapy for familial hypercholesterolaemia: problems, progress, and perspectives. Int Arch Med. 2010; 3:36. [ Links ]
73. Martínez L, Ordóñez M.L, Letona R, Sumano VO, Guerra MM, Tusié-Luna MT, Aguilar-Salinas CA. Hipercolesterolemia familiar homocigota por la mutación c2271delT del gen del receptor LDL, detectada únicamente en mexicanos. Gac Med Mex. 2011; 147:394-8. [ Links ]
74. Riba L. Genes implicados en las formas monogénicas de la hipercolesterolemia familiar. Rev Endocrinol Nutr. 2008; 16(1): 24-31. [ Links ]
75. Schaefer JR. Unraveling hyperlipidemia type III (dysbetalipoproteinemia), slowly. Eur J Hum Genet. 2009; 17(5):541–542. [ Links ]
76. Henneman P, Sman-de Beer F, Moghaddam PH, Huijts P, Stalenhoef A, Kastelein JJ, Duijn C, Havekes LM, Frants RR, Dijk KW, Smelt A. The expression of type III hyperlipoproteinemia: involvement of lipolysis genes. Eur J Hum Genet. 2009; 17(5):620-628. [ Links ]
77. Furgione A, Sánchez D, Scott G, Luti Y, Arraiz N, Bermúdez V, Velasco M. Dislipidemias primarias como factor de riesgo para la enfermedad coronaria. Rev Latinoam hipertens. 2009; 4:18-25. [ Links ]
78. Himbergen T, Otokozawa S, Matthan NR, Schaefer EJ, Buchsbaum A, Masumi A, Tits L, Graaf J, Stalenhoef AF. Familial Combined Hyperlipidemia is Associated with Alterations in the Cholesterol Synthesis Pathway. Arterioscler Thromb Vasc Biol. 2010; 30(1):113. [ Links ]
79. Cabre A, Lazaro I, Cofan M, Jarauta E, Plana N,Garcia-Otin AL, Ascaso JF, Ferre R, Civeira F, Ros E, Masana L. FABP4 plasma levels are increased in familial combined hiperlipidemia. J Lipid Res. 2010; 51(5): 1173–1178. [ Links ]
80. De Abajo OS. Epidemiología, definición, clasificación, despistaje y diagnóstico de las dislipemias. SEMERGEN. 2009; 35(3):3-9. [ Links ]
81. Arráiz N, Pacheco M, Prieto C, Escalona C, Mujica A, Mujica E, Chacín M, Añez R, Bello L, Roque W, Toledo A, Bermúdez V. Mutaciones en la región codificante del dominio de unión de la apolipoproteina B-100: diagnóstico de apolipoproteína B defectuosa familiar. Rev Latinoam Hipertens. 2011; 6:30-34. [ Links ]
82. Konrad RJ, Troutt JS, Cao G. Effects of currently prescribed LDL-C-lowering drugs on PCSK9 and implications for the next generation of LDL-C-lowering agents. Lipids Health Dis. 2011; 10:38. [ Links ]
83. Essalmani R, Susan-Resiga D, Chamberland A, Abifadel M, Creemers JW, Boileau C, Seidah NG, Prat A. In Vivo Evidence That Furin from Hepatocytes Inactivates PCSK9. J Biol Chem. 2011; 286(6):4257-4263. [ Links ]
84. Soca P. Dislipidemias. ACIMED. 2009; 20(6): 265-273. [ Links ]
85. Elisaf M, Tsimihodimos V. Editorial: Secondary Dyslipidemias. Open Cardiovasc Med J. 2011; 5: 22-3. [ Links ]
86. Shafiq N, Singh M, Kaur S, Khosla P, Malhotra S. Dietary treatment for familial hypercholesterolaemia. Cochrane Database Syst Rev. 2010;1: CD001918. [ Links ]
87. Jenkins DJ, Wong JM, Kendall CW, Esfahani A, Ng VW, Leong TC, Faulkner DA, Vidgen E, Greaves KA, Paul G, Ng VW, Leong TC. The effect of a plant-based low-carbohydrate ("Eco-Atkins") diet on body weight and blood lipid concentrations in hyperlipidemic subjects. Arch Intern Med. 2009; 169(11):1046-54. [ Links ]
88. Jones PJ, MacDougall DE, Ntanios F, Vanstone CA. Dietary phytosterols as cholesterol-lowering agents in humans. Can J Physiol Pharmacol. 1997; 75:217-227. [ Links ]
89. Sudhop T, Von Bergmann K. Cholesterol absorption inhibitors for the treatment of hypercholesterolaemia. Drugs. 2002; 62:2333-47. [ Links ]
90. Peña S. Flora intestinal, probióticos, prebióticos, simbióticos y alimentos Novedosos. Rev Esp Enferm Dig (Madrid). 2007; 99(11):653-658. [ Links ]
91. Taranto MP, Médici M, Valdez GF. Alimentos funcionales probióticos. Rev química viva. 2005; 1: 26-34. [ Links ]
92. Liong MT, Shah NP. Optimization of Cholesterol Removal by Probiotics in the Presence of Prebiotics by Using a Response Surface Method. APPL. Envirion Microbiol. 2005; 71(4):1745-1753. [ Links ]
93. Mann GV, Spoerry A. Studies of a surfactant and cholesteremia in the Maasai. Am J Clin Nutr. 1974; 27:464-469. [ Links ]
94. Xie N, Cu Yi, Yin YN, Zhao X, Yang JW, Wang ZG, Fu N, Tang Y, Wang XH, Liu XW, Wang CL, Lu FG. Effects of two Lactobacillus strains on lipid metabolism and intestinal microflora in rats fed a high-cholesterol diet. BMC Complement Altern Med. 2011; 11:53. [ Links ]
95. Kim Y, Whang JY, Whang KY, Oh S, Kim SH. Characterization of the Cholesterol-Reducing Activity in a Cell-Free Supernatant of Lactobacillus acidophilus ATCC 43121. Biosci Biotechnol Biochem. 2008; 72(6):1483-1490. [ Links ]
96. Bailén, MR. Administración de estatinas durante la fase aguda del síndrome coronario agudo. Med Intensiva. 2010; 34(1):56-63. [ Links ]
97. Hernández-Pérez F, Herrera-Arellano A. Tratamiento de la Hipercolesterolemia con Hibiscus sabadariffa. Rev Med Inst Mex Seguro Soc. 2011; 49(5):469-480. [ Links ]
98. Rodríguez L, Alcalá J, Refoyo E. Beneficios de la terapia combinada en la hiperlipemia. Rev Esp Cardiol. 2006; 6(Supl G):63-71. [ Links ]
99. Sjouke B, Kusters DM, Kastelein JJ, Hovingh GK. Familial Hypercholesterolemia: Present and Future Management. Curr Cardiol Rep. 2011; 13:527-536. [ Links ]
100. Ros E. Inhibición de la absorción intestinal del colesterol: nueva diana terapéutica en la reducción de la colesterolemia. Clin Invest Arterioscl. 2003; 15:261-75. [ Links ]
101. Madrigal J, Herrera A, Lemus H. Efectos de la combinación de ezetimiba más estatinas sobre los lípidos en pacientes mexicanos Med Int Mex. 2007; 23(4):280-85. [ Links ]
102. Kastelein JJ, Akdim F, Stroes ES, Zwinderman AH, Bots ML, Stalenhoef AF, Visseren FL, Sijbrands EJ, Trip MD, Stein EA, Gaudet D, Duivenvoorden R, Veltri EP, Marais AD, de Groot E. Simvastatin with or without ezetimibe in familial hypercholesterolemia. N Engl J Med. 2008; 358(14):1431-1443. [ Links ]
103. Davidson MH, McGarry T, Bettis R. Ezetimibe coadministered with simvastatin in patients with primary hypercholesterolemia. J Am Coll Cardiol. 2002;40(12):2125-2134. [ Links ]
104. Kerzner B, Corbelli, J, Sharp S, Lipka LJ, Melani L, LeBeaut A, Suresh R, Mukhopadhyay P, Veltri EP. Efficacy and safety of ezetimibe coadministered with lovastatin in primary hypercholesterolemia. Am J Cardiol. 2003;91:418-24. [ Links ]
105. Melani L, Mills R, Hassman D, Lipetz R, Lipka L, LeBeaut A, Suresh R, Mukhopadhyay P, Veltri E. Efficacy and safety of ezetimibe coadministered with pravastatin in patients with primary hypercholesterolemia: a prospective, randomized, double-blind trial. Eur Heart J. 2003;24:717-28. [ Links ]
106. De Bari O, Neuschwander-Tetri BA, Liu M, Portincasa P, Wang DQ. Ezetimibe: Its Novel Effects on the Prevention and the Treatment of Cholesterol Gallstones and Nonalcoholic Fatty Liver Disease. J Lipids. 2012; 2012: 302847. [ Links ]

