










Servicios Personalizados
Revista
Articulo
 Español (pdf)
Español (pdf)
 Artículo en XML
Artículo en XML Referencias del artículo
Referencias del artículo
 Enviar artículo por email
Enviar artículo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO -
 Accesos
Accesos
Links relacionados
-
 Similares en
SciELO
Similares en
SciELO
Compartir

 Permalink
PermalinkRevista mexicana de ciencias farmacéuticas
versión impresa ISSN 1870-0195
Rev. mex. cienc. farm vol.42 no.1 Ciudad de México ene./mar. 2011
Trabajos científicos
Desarrollo y validación de un método analítico por cromatografía de líquidos de alta resolución para la determinación de indometacina en crema
Development and validation of an analytical method by high performance liquid chromatography for determination of indomethacin in cream
María Luisa Vázquez R., Alejandra Hernández L., Berta Retchkiman C.
Departamento de Sistemas Biológicos, Universidad Autónoma Metropolitana-Xochimilco
Correspondencia:
M en C Ma. Luisa Vázquez R.
Departamento de Sistemas Biológicos
Universidad Autónoma Metropolitana-Xochimilco
Calzada del Hueso 1100, Colonia Villa Quietud
Delegación Coyoacán C.P. 04960, México D.F., México
Tel. 5483 7511 Fax 5483 7511, 5483 7237
e-mail: lramirez@correo.xoc.uam.mx
Fecha de recepción: 11 de noviembre de 2010.
Fecha de recepción de modificación: 7 de febrero de 2011.
Fecha de aceptación: 21 de febrero de 2011.
Resumen
Se desarrolló un método analítico rápido y sencillo por Cromatografía de Líquidos de Alta Resolución (CLAR) con detección UV para la determinación de indometacina en crema al 2.5%. Se probaron diferentes condiciones para la cuantificación, encontrando mejores resultados con una columna L, (Supelcosil LC18, 250 mm x 4.6 mm, 5 μm), una fase móvil compuesta por metanol y solución amortiguadora de fosfatos pH 7.1 (70:30), una velocidad de flujo de 1.0 mL/min y una longitud de onda de detección de 254 nm. El método es rápido y lineal en un intervalo de concentraciones de 10 a 50 μg/mL (r2 = 0.999), exacto (IC 100.32±4.5%) y preciso (CV< 1.5%) resultando adecuado para la cuantificación del fármaco como método de control de calidad.
Palabras clave: indometacina, CLAR, ingredientes activos farmacéuticos.
Abstract
An easy, quick and completely automatizated analytical method was developed by High Performance Liquid Chromatography (HPLC) with UV detection for quantitative determination of Indomethacin in 2.5% cream. Different conditions for its determination were tested, finding the best conditions using a L column1 (Supelcosil LC18, 250 mm x 4.6 mm, 5 μm), a movil phase with metanol and phosphate buffer pH 7.1 (70:30), flow speed 1.0 mL/min and detection lambda of 254 nm. The method is quick and linear from 10 to 50 μg/mL ((r2 = 0.999), accurate (IC 100.32±4.5%) and adequate precision (CV< 1.5%), and it results in an adequate analytical method to quantification of Indomethacin in a quality control test.
Key words: indomethacin, HPLC, pharmaceuticals.
Introducción
La indometacina [Ácido 1-(4-clorobenzoil)-2-metil-5metoxi-1H-indol-3-acético] -metilindol-3-il] acético; Ácido (1-(4-clorbenzoil)-5-metoxi-2-metilindo-3-leacetico)] es un antiinflamatorio no esteroideo, analgésico y antipirético. Su efecto se basa en la inhibición de la síntesis de prostaglandinas en tejidos periféricos y a la inhibición de la migración leucocitaria a las áreas inflamadas. Se usa frecuentemente en el tratamiento de los síntomas de la artritis reumatoide, osteoartritis y espondilitis anquilosante. La vía de administración usualmente es oral, pero puede administrarse por vía intravenosa, vía rectal o tópica1.
Se han utilizado una gran variedad de técnicas analíticas para la cuantificación de indometacina como materia prima y en preparaciones farmacéuticas principalmente tabletas y cápsulas, por ejemplo, titulaciones ácido-base2, espectrofotometría3-5, espectrofluorometría6, electroforesis capilar7, densitometría8 y cromatografía de líquidos de alta resolución (CLAR).9-14
En la mayoría de los métodos publicados para cuantificar indometacina en plasma también se determina por CLAR.15-18
La metodología analítica de la FEUM 9a edición19, incluye en sus monografías el análisis por CLAR, para indometacina materia prima y un método por espectrofotometría UV, para la determinación de indometacina en tabletas utilizando cloruro de metileno como disolvente final.
La valoración de la suspensión de indometacina se lleva a cabo con un procedimiento de extracción utilizando una mezcla de agua y éter, posteriormente, se disuelve una alícuota de la fase etérea, en una mezcla de metanol:ácido acético glacial (99:1) determinando la absorbancia de la solución a 320 nm.
La USP 3220 recomienda un método por CLAR para indometacina materia prima utilizando una columna analítica conteniendo empaque L1 (4mm x 30cm, 10 μm). La cuantificación de indometacina en cápsulas y supositorios se determina por un método espectrofotométrico a 320 nm con cloruro de metileno como disolvente. Para las cápsulas de liberación extendida y la suspensión de indometacina se utiliza un método por CLAR con una columna L1. Las muestras se disuelven en ácido fosfórico diluido utilizando un sonicador, para ser inyectadas posteriormente en el cromatógrafo.
En diversos artículos científicos se ha reportado metodología analítica por CLAR para la determinación de indometacina con detección ultravioleta. Khuhawar y col. desarrollaron un método para cuantificar isoniazida y pirazinamida en tabletas e indometacina en cápsulas. La columna utilizada es YMC-ODS (150mm x 4.6 mm d.i), con una fase móvil agua:metanol:tetrahidrofurano (59:39:2) y una velocidad de flujo de 2 mL/min a una longitud de onda de 328 nm. El rango de concentraciones estudiadas fue de 10.7 a 64.08 μg/mL.11
Para la cuantificación de indometacina en gel por CLAR Nováková y colaboradores utilizaron varias columnas para el desarrollo de este método, siendo la mejor Zorbax SB-Phenyl (75 mm x 4.6 mm, 3.5 μm) con la cual se eliminaban las interferencias del placebo, una fase móvil compuesta de acetonitrilo:ácido fosfórico 0.2% (50:50) y una velocidad de flujo de 1.2 mL/min utilizando para la determinación una longitud de onda de 237 nm.10
El desarrollo de la na notecnología implica la formación de partículas lipídicas sólidas de tamaño nanoscópico que contienen al fármaco de interés, para mejorar las propiedades de solubilidad e incrementar el área superficial efectiva, entre otros aspectos, lo que las convierte en una excelente alternativa a sistemas de liberación como emulsiones y liposomas.21 Estos sistemas de liberación ahora se enfrentan al reto, de desarrollar conjuntamente ensayos validados que permitan detectar y cuantificar el fármaco contenido en las nanopartículas en productos terminados.22,23
Para el caso específico de Indometacina, el desarrollo de la nanotecnología ha empleado esta metodología para elaborar principalmente cremas de aplicación tópica lo que facilita la absorción de la indometacina a través de la epidermis, sobre todo en los tejidos que rodean las articulaciones donde se requiere la acción del fármaco.
Es importante resaltar que aunque existe un gran número de métodos por CLAR para cuantificar indometacina, no existe un método para su cuantificación en la forma farmacéutica de crema (conteniendo nanopartículas). Para alcanzar este propósito, en este artículo se presenta el desarrollo de un método por CLAR simple, sencillo y rápido para la determinación de indometacina en este tipo de formas farmacéuticas.
Material y método
Reactivos
El estándar de referencia de Indometacina fue proporcionado por FEUM. El fosfato monobásico de potasio, el fosfato dibásico de sodio y el ácido ortofosfórico fueron grado reactivo (Baker). El metanol fue grado cromatográfico (Merck). El agua utilizada fue grado cromatográfico preparada con Direct Q3 Millipore.
Equipo
Para llevar a cabo todos los análisis se utilizó un sistema cromatográfico Perkin Elmer modelo LC-95 equipado con una bomba cuaternaria, un inyector manual y un detector UV/ visible de longitud de onda variable 553. El equipo cuenta con un sistema de inyección de volumen constante calibrado a 20 μL. La separación de la indometacina se llevó a cabo con una columna L1 (Supelcosil LC18), de 250 mm x 4.6 mm y 5 μm de tamaño de partícula. Se tenía acoplado un integrador Waters 745 Data Module.
Condiciones cromatográficas
Se probaron diferentes mezclas para la fase móvil, encontrando que la más adecuada fue metanol: solución amortiguadora de fosfatos pH 7.1, 70:30 con una velocidad de flujo de 1 mL/min y la detección a una longitud de onda de 254 nm.
Preparación del estándar y curva estándar
La solución del estándar de referencia para el análisis de la crema de indometacina, se preparó disolviendo 50 mg de indometacina FEUM en metanol y llevando a un aforo a 100 mL con el mismo disolvente. Posteriormente, se realizó una curva de calibración con un rango de concentraciones de 10 a 50 μg/mL haciendo la dilución final con fase móvil.
Preparación de la muestra
Se pesaron exactamente 800 mg de crema de indometacina equivalente a 20 mg de principio activo, se le adicionó 50 mL de metanol y se sometió a sonicación por 15 minutos, posteriormente se aforó a 100 mL con metanol y se filtró utilizando un filtro Millipore de 0.45 μm. Las diluciones finales para la curva de calibración con la muestra se llevaron a cabo con fase móvil.
Método de validación
La validación del método se realizó con base en las guías de ICH y los requerimientos farmacéuticos de fuente farmacopéica. Una parte importante de la validación, es la adecuabilidad del sistema, para lo cual se calcularon el número de platos teóricos, la asimetría del pico, resolución y área bajo la curva. Todos los análisis estadísticos fueron realizados con el software SPSS versión 19.
Linealidad y precisión
La linealidad del sistema se determinó con el coeficiente de correlación en un rango de concentraciones de 10 a 50 μg/ mL (33.34-166.67%), utilizando cinco curvas de calibración del estándar disuelto en la fase móvil metanol: solución amortiguadora pH 7.1 (70:30). La precisión del sistema fue evaluada con el coeficiente de variación utilizando nueve muestras de la concentración correspondiente al 100%.
Para la linealidad del método se analizaron cinco curvas de calibración con la crema de indometacina en un intervalo de concentraciones de 10 a 50 μg/mL. Para llevar a cabo el análisis del coeficiente de correlación y el análisis de varianza de la regresión, la variable de respuesta considerada es el área bajo la curva en función de la concentración.
Se evalúa el análisis de regresión para el porcentaje de recuperación obtenido bajo el tratamiento de las muestras como se ha descrito.
Exactitud y precisión
La exactitud del método se determinó con 30 muestras de crema a tres rangos de concentración 24, 30 y 36 μg/mL (80,100 y 120 %), comparados contra la concentración nominal de muestras control.
La precisión fue evaluada por el análisis de muestras de diferentes concentraciones en diferentes días de análisis. Y se evaluó a través de los resultados globales en todas las pruebas de validación.
Reproducibilidad
Se analizaron por triplicado muestras de tres concentraciones conocidas de indometacina crema, en tres días. Se evaluó la posible diferencia a través de un análisis de varianza de dos criterios de clasificación con interacción. Y se reporta el coeficiente de variación inter-día y general.
Selectividad y límite de detección
La selectividad del método se verificó por comparación de la muestra del preparado farmacéutico y la solución estándar. El límite de detección se determinó con el método de promedio señal-ruido.
Estabilidad
La estabilidad de la muestra en refrigeración a 4 °C fue evaluada por un periodo de 3 meses.
Resultados y discusión
Para la determinación de la indometacina en crema, se eligió CLAR, con detección por espectrofotometría como un método sencillo rápido y efectivo para la determinación del principio activo.
Se probaron diferentes condiciones para el desarrollo de este método, utilizando metanol, solución amortiguadora de fosfatos y acetonitrilo en diferentes combinaciones. La fase móvil final fue metanol: fosfatos pH 7.1.
El tiempo de retención obtenido para Indometacina bajo las condiciones descritas fue de 1.78 min para el estándar y 1.77 min para la muestra en la presentación en crema (Figura 1).

Se lograron obtener valores dentro de las especificaciones para la adecuabilidad del sistema (Tabla 1).

La linealidad del sistema fue estimada por análisis de regresión utilizando las cinco curvas de calibración elaboradas con solución de estándar en concentraciones de 10 a 50 μg/mL, obteniendo el siguiente modelo ajustado:
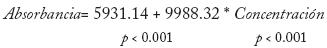
El cual demostró ser significativo (p < 0.0001), con r = 0.9998 y r2 = 0.9995. Tanto el intercepto como la pendiente son significativamente diferentes de cero (p<0.001 en ambos casos). análisis de varianza mostró que el modelo es adecuado para la representación de los datos, como se muestra en la Tabla 2 con p < 0.0001. La gráfica de valores observados y ajustados se muestra en la Figura 2.

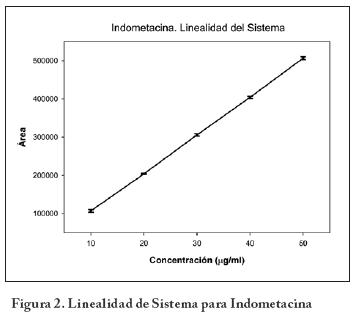
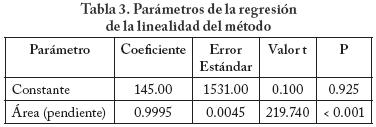
Para la linealidad del método se analizaron cinco curvas de calibración con la crema de indometacina en un intervalo de concentraciones de 10 a 50 μg/mL. Se realizó la regresión entre la cantidad adicionada y la cantidad recuperada para evaluar el manejo de la muestra y el intervalo de trabajo.
Los parámetros de la regresión se muestran en la Tabla 3, y se concluye que el método es adecuado para el manejo de la muestra ya que el intercepto es estadísticamente igual que cero, mientras que la pendiente es estadísticamente igual a 1, por lo que la recuperación de las cantidades agregadas se acerca a 100%. La regresión concluye con un coeficiente de correlación de 0.9999 y un valor de predicción con 95% de confianza de 0.9999, lo cual es significativo para el modelo de recuperación de las cantidades agregadas, logrando un método lineal.
La exactitud fue evaluada en 30 muestras preparadas con cantidades agregadas de 23.95, 30.2045 y 34.82 μg/mL, se obtuvo una media de 100.329 % ± 1.52% (± desviación estándar), con intervalo de confianza de 99.76% a 100.90%, lo que demuestra que el método es exacto. Adicionalmente se realiza un análisis de varianza para evaluar la posible diferencia entre concentraciones, el cual se muestra en la Tabla 4, y se concluye que no existe diferencia entre concentraciones (p = 0.462), por lo que el método es exacto para las concentraciones evaluadas.

La reproducibilidad se estima a través de un análisis de varianza de dos criterios de clasificación con interacción al evaluar tres concentraciones diferentes en tres días de análisis. El coeficiente de variación intradía fue de 1.07%, 1.00% y 1.25%. El coeficiente de variación global para los datos fue de 1.09%.
A través de un análisis de varianza se estima la posible diferencia entre días y entre concentraciones, que se muestra en la Tabla 5. No se encuentran diferencias entre días o entre concentraciones. Igualmente la interacción no fue significativa. La conclusión es que el método es reproducible.

La estabilidad de la muestra se evaluó en un periodo de tres meses, en una serie de 4 muestras, las cuales se mantuvieron en refrigeración a 4°C, obteniendo una diferencia de 0.98% ± 0.13% en el periodo establecido y sin que apareciera ninguna señal adicional en los cromatogramas.
Conclusiones
De acuerdo a los criterios de ICH y de las guías de validación utilizadas, el método cumple con criterios de sistema y de método (linealidad, reproducibilidad, exactitud), por lo que se concluye que se logra obtener un método con precisión adecuada de valores pequeños de coeficientes de variación y con términos independientes en días de análisis y a diferentes concentraciones. Las muestras son estables al ser mantenidas en refrigeración a 4°C.
El método desarrollado es adecuado para su uso en el laboratorio de control de calidad para el análisis de rutina de Indometacina Crema, considerando que es una formulación nanoparticulada.
Referencias
1. Vadem Intl. UBM Medica España. Edición 2010. [ Links ]
2. Eur Pharm Sixth edition 2007 Convention on the Elaboration of a European Pharmacopoeia (European treaty series No. 50). Published: Directorate for the Quality of Medicines & Health Care of the Council of Europe.
3. Adegoke O.A.,Idowu O.S., Olaniyi A.A. 2006. Novel colorimetric assay of indomethacin using 4-carboxyl-2,6-dinitrobenzene diazonium ion. Acta Pharm, 56(2):189-202. [ Links ]
4. Nagaraja P., Vasantha R.A., Yathirajan H.S.2003. Sensitive spectrophotometric method for the determination of indomethacin in capsules. J Pharm Biomed Anal, 31(3):563-569. [ Links ]
5. Aman T.,Naureen F., Kazi A.A., Khan I.U., Kashmiri M. A. 2002. Ammonium molybdate as a spectrophotometric reagent for the determination of indomethacin in pure and pharmaceutical preparations. Anal Let, 35(6):1007-1020. [ Links ]
6. Pinto P., Saraiva M.L., Santos J., Lima J. 2005. A pulsed sequential injection analysis flow system for the flourimetric determination of indomethacin in pharmaceutical preparations. Anal Chim Acta, 539:173-179. [ Links ]
7. Makino K., Yano T., Maiguma T., Teshima D., Sendo T., Itoh Y., Oishi R. 2003. A rapid and simultaneous determination of several analgesic antiinflammatory agents by capillary zone electrophoresis. Ther Drug Monit, 25(5): 574-580. [ Links ]
8. Krzek J.,Starek M. 2001. Simultaneous densitometric determination of indomethacin and its degradation products, 4-chlorobenzoic acid and 5-methoxy-2-methyl-3-indoleacetic acid, in pharmaceutical preparations. J AOAC Int, 84(6):1703-1707. [ Links ]
9. Abdel-Hamid M.E., Novotny L., Hamza H. 2001. Determination of diclofenac sodium, flufenamic acid, indomethacin and ketoprofen by LC-APCI-MS. J Pharm Biomed Anal, 24:587-594. [ Links ]
10. Nováková L., Matysová L., Havlíková L., Solich P. 2005.Development and validation of HPLC method for determination of indomethacin and its two degradation products in topical gel. J Pharm Biomed Anal, 37:899-903. [ Links ]
11. Khuhawar M.Y., Rind F.M.A., Rajper A.D. 2005. High-Performance liquid chromatographic determination of isoniazid, pyrazinamide, and indomethacin en pharmaceutical preparations. Acta Chromatogr, 15:269-275. [ Links ]
12. Martínez-Algaba C., Escuder-Gilabert L., Sagrado S., Villanueva-Camañas R.M., Medina-Hernández J.M. 2004. Comparison between sodium dodecylsulphate and cetyltrimethylammnoium bromide as mobile phases in the micellar liquid chromatography determination of non-steroidal anti-inflammatory drugs in pharmaceuticals. J Pharm Biomed Anal, 36(2):393-399. [ Links ]
13. Xueqi L., Ling L., Lianqiu L. 2007. Determination the Content of Indometacin Enteric-Coated Tablets by HPLC. Chin Pharm, 3:27. [ Links ]
14. Zhang Y., Zhang Z., Qi G., Sun Y., Wei Y., Ma H. 2007. Detection of indomethacin by high-performance liquid chromatography with in situ electrogenerated Mn (III) chemiluminescence detection. Anal Chim Acta, 582(2):229-234. [ Links ]
15. Al Za'abi M.A., Dehghanzadeh G.H., Norris R.L., Charles B.G. 2006. A rapid and sensitive microscale HPLC method for the determination of indomethacin in plasma of premature neonates with patente ductus arteriousus. J Chromatogr B-anal Tech Biomed Sci, 830(2):364-367. [ Links ]
16. Boon V., Glass B., Nimmo A. 2006. High-Performance Liquid Chromatographic Assay of Indomethacin in Porcine Plasma with Applicability to Human Levels. J Chromatogr Sci, 44(1):41-44. [ Links ]
17. Johnson AG., Ray J.E. 1992. Improved high-performance liquid chromatographic method for the determination of indomethacin in plasma. Ther Drug Monit, 14(1):61-65. [ Links ]
18. Sato J., Amisuka T., Umetsu M., Ito K. 1997. Simple, rapid and sensitive method for the determination of indomethacin in plasma by high-performance liquid chromatography with ultraviolet detection. J Chromatogr B. Biomed Sci App, 692(1):241-244. [ Links ]
19. FEUM. 2008. 9a. Edición. Secretaría de Salud y Comisión Permanente de la Farmacopea de los Estados Unidos Mexicanos. [ Links ]
20. USP-NF2009 (USP 32 NF 27). United States Pharmacopeial Convention, Inc., meeting at Washington, D.C. Prepared by the Committee of Revision and published by the Board Trustees. [ Links ]
21. Müller RH., Máder K., Gohla S., 2000: Solid lipid nanoparticles (SLN) for controlled drug delivery - a review of the state of the art. Eur J Pharm Biophar, 50, 161-177. [ Links ]
22. Carlucci AM., Bregni C., 2009. Productos nanotecnológicos de aplicación en farmacoterapia. Lat Am J Pharm, 28(3), 470-477. [ Links ]
23. Wechsler J.2006.: Los nanomateriales plantean nuevos desafíos. Pharm Technol (en Español), Sep/Oct 2006, 62-66. [ Links ]

