











 nova página do texto(beta)
nova página do texto(beta) Inglês (pdf)
Inglês (pdf)
 Artigo em XML
Artigo em XML Referências do artigo
Referências do artigo
 Enviar este artigo por email
Enviar este artigo por email Citado por SciELO
Citado por SciELO  Similares em
SciELO
Similares em
SciELO 
 Permalink
Permalink1. Introduction
Peptide nucleic acids (PNAs), introduced by Nielsen and coworkers in 1991 are DNA/RNA analogues (Nielsen, Egholm, Berg, & Buchardt, 1991), whereby N-2-aminoethylglycine repeating units replace the sugar-phosphate backbone and the polyamide chain is linked to nucleobases covalently through a carboxymethyl spacer. The attached nucleobases are natural and so through Watson-Crick base pairing are able to bind to complementary DNA/RNA (Egholm et al., 1993). Since the PNA backbone does not contain any charged phosphate group, the absence of electrostatic repulsion gives rise to a stronger binding between PNA/DNA strands than that of DNA/DNA duplex. Consequently, the thermal stability of PNA/DNA duplexes as compared to natural DNA/DNA double helix of the same length is higher. Moreover, unlike DNA/DNA, PNA/DNA duplex is much less affected by medium with high ionic strength medium. It was recently found in lieu of finding relationship of PNA to the origin of life that PNA-like materials are present in cyanobacteria (Banack et al., 2012).
The two strands in PNA/DNA hybrid can be fashioned in either parallel or antiparallel orientation but the later mode grabs higher stability because of the achiral backbone of PNA. The stability of the PNA/DNA duplexes is also found to be highly sensitive to the existence of a single mismatched base pair. PNA probes are very sequence-selective and advanced to DNA probes in single-base mismatch recognition. As enzymes are substrate specific, the recognition of neutral backbone of PNA is not easy by either nucleases or proteases, making them potentially resistant to enzymatic degradation and their stability over wide pH range.
The major drawbacks like poor water solubility, inefficient cellular uptake, self-aggregation and ambiguity in directionality of binding restricts its applications within medicine, diagnostics, molecular biology, etc. One approach to improve cellular uptake is to conjugate PNAs to a wide variety of ligands, such as artificial nucleases, peptides, intercalators or fluorescent reporter groups in order to combine the favourable properties of both entities in a single construct. In most of these conjugates, the attachment of ligand is either to the C- or N-terminal end of the PNA. Over the years, a number of backbone-modified PNAs have been prepared to study the effect of introducing chirality, charge or steric bulk on their properties such as hybridization or solubility. The application of a suitably protected thiol modified PNA monomer would give access to a PNA oligomer containing a sulfhydryl group suitable for post-assembly conjugation methods (De Koning et al., 2002; De Koning, Van Der Marel, & Overhand, 2003; De Koning, Filippov, Van der Marel, Van Boom, & Overhand, 2003; De Koning, Filippov, Van der Marel, Van Boom, & Overhand, 2004; De Koning et al., 2006; Dose & Seitz, 2005; Goodwin, Holland, Lay, & Raney, 1998; Liu & Balasubramanian, 2000).
2. Experimental
2.1. General procedure
Melting points were determined on a sulphuric acid bath and are uncorrected. The IR spectra were recorded on a Perkin Elmer model 2000 FT-IR spectrophotometer by making KBr discs for solid samples and chloroform film for viscous samples. The 1H NMR and 13C NMR spectra were recorded on Bruker Avance 300 spectrometer and Bruker-500 using TMS as internal standard. The chemical shift values are on S scale and the coupling constant values (J) are in Hz. EI mass spectra were recorded on Agilent-6210 ESI-TOF. Analytical TLCs were performed on pre-coated Merck silica gel 60F254 plates with fluorescence indicator; the spots were detected by viewing under UV light or by iodine chamber. Column chromatography was carried out using silica gel (100-200 mesh). U.V. data was recorded on Shimadzu UV-2501PC UV-vis spectrophotometer. All other chemicals used were purchased either from S. D. Fine Chemicals, Spectrochem, India or Aldrich Chemical Co., USA and used without further purification.
2.2. Synthesis of S-p-methoxybenzyl cysteine methylester hydrochloride (2) (Richter, Marsters, & Gadek, 1994)
A solution of 4-methoxybenzyl chloride (5.8 g, 37 mmol) in dichloromethane (250 mL) was added dropwise to a solution of L-cysteine (5g, 37mmol) in trifluoro acetic acid/dichloromethane (45 mL/250 mL), over a period of 1 h at 0°C. The reaction mixture was stirred for 2 h at room temperature. Then methanol (150 mL) and water (500 mL) were added, the layers were separated and the organic layer was re-extracted with water (100 mL). The aqueous layer was washed with DCM, filtered and solvents were removed in vacuo. The residue was dissolved in water, brought to pH 6 with 10% NaHCO3 and the product was recrystallized from methanol/water (3:1). The obtained compound (5 g) was dissolved in anhydrous methanol (100 mL) and thionyl chloride (6 mL) was added dropwise at 0 ° C. The reaction mixture was stirred for 3 h at room temperature. Methanol was then evaporated and the residue was washed with diethyl ether. It was obtained as white solid (80%); melting point: 80 °C; 1H NMR (CDCl3, 300MHz): δ 2.88-2.91 (m, 1H, Ha), 2.98-3.04 (m, 1H, Hb), 3.77 (s, 2H, H-5), 3.78, 3.82 (2 x s, 6H, H-1 and H-10), 4.12 (m, 1H, H-3), 6.88 (d, J =8.7Hz, 2H, 2 x H-8), 7.28 (d, J =8.7Hz, 2H, 2 x H-7); 13C NMR (CDCl3,125MHz): δ 31.62 (C-4), 35.74 (C-5), 52.66 (C-1), 53.27 (C-3), 55.97 (C-10), 114.69 (2 x C-8), 130.38 (C-6), 131.11 (2 x C-7), 159.24 (C-9) and 169.52 (C-2); IR (KBr) υmax 3412.57 (NH2 str), 2840.36, 2637.14, 1745.85 (C=O), 1610.29, 1581.69, 1510.70, 1439.71, 1324.76, 1304.90, 1247.67, 1204.88, 1176.08, 1150.57, 1106.61, 1059.79, 1034.70, 995.41, 930.54, 870.01, 830.93, 777.10, 684.89 and617.09 cm-1;UV (methanol) λmax 278 nm; HRMS: C12H18O3NSCl [M]+ 291.2902.
2.3. Synthesis of N4-benzyloxycarbonyl cytosine-1-yl-acetic acid (3) (Dueholm et al., 1994)
Benzyloxycarbonyl chloride (5.2 mL, 36mmol) was added dropwise to a suspension of cytosine (2g, 18mmol) in anhydrous pyridine (100 mL) at 0 oC. The mixture was stirred overnight and, subsequently, the pyridine suspension was evaporated to dryness. Water (20 mL) was added and pH was adjusted to 1 with aqueous 4 M HCl. The resultant white precipitate was filtered off, washed with water and partially dried. The wet precipitate was boiled with absolute ethanol (50 mL) for 10 min, cooled to 10 °C, filtered, washed thoroughly with ether to yield compound 6 in 85% yield. Ethyl bromoacetate (0.5 mL, 4.1 mmol) was then added to a suspension of N 4-benzyloxycarbonyl cytosine (1 g, 4.1 mmol) and K2CO3 (570 mg, 4.1 mmol) dissolved in dimethyl formamide (10 mL). The reaction mixture was stirred overnight at room temperature. The solution was then filtered and evaporated to dryness. The solid residue was treated with water (4 mL) and 4 M HCl (0.2 mL), stirred for 15 min at 0 °C, filtered and washed with water. Water (2.5 mL) was added to N 4-benzyloxycarbonyl cytosine-1-yl-acetic acid ethyl ester (800 mg, 2.4 mmol) and 2 M NaOH (3 mL) and stirred for 2 h. The reaction mixture was cooled to 0 °C and filtered. The compound was precipitated by the addition of 4 M HCl (2 mL). The compound was obtained by recrystallization with methanol as white solid (60%); melting point: 272 °C (Literature value = 266-274 °C); 1H NMR (CDCl3, 300MHz): δ 4.54 (s, 2H, H-14), 5.19 (s, 2H, H-9), 7.06 (d, J =6.8Hz, 1H, H-5), 7.35-7.40 (m, 5H, C6H5), 8.00 (d, J=6.6Hz, 1H, H-4); 13C NMR (CDCl3, 75 MHz): δ 50.54 (C-14), 66.53 (C-9), 94.03 (C-5), 127.95 (2x C-11), 128.17 (C-13), 128.49 (2x C-12), 135.96 (C-10), 150.42 (C-4), 153.18, 155.03 (C-2 and C-8), 163.31, 169.37 (C-6 and C-15); IR (KBr) υmax: 3423.87 (NHCO), 3143.94, 2953.22, 2524.06, 1759.05 (NHCO), 1712.63 (COOH), 1678.72 (C=O), 1629.46, 1582.61, 1517.58, 1454.32, 1416.24, 1371.21, 1332.40, 1248.15, 1215.41, 1069.28, 1007.24, 981.10, 901.54, 802.38, 780.06, 738.75, 694.29, 634.79, and 607.86 cm-1; UV (methanol) λmax: 298nm; HRMS: C14H13O5N3 [M]+ 303.6239.
2.4. Synthesis of [(methoxymethylcarbamoyl)-methyl]-carbamic acid tert-butyl ester (4)
Hydroxybenzotriazole (7g, 45.7mmol) was added to a solution of boc-glycine(5 g, 28.6 mmol) in anhydrous dimethyl formamide (50 mL) followed by the addition of N,O-dimethylhydroxylamine hydrochloride (4.5 g, 45.7 mmol), DIPEA (7.9 mL, 45.7 mmol) and DIPC (9 mL, 57.1 mmol). The reaction mixture was stirred overnight at room temperature. The solution was poured into water (100 mL) and then filtered. The compound was extracted with ethyl acetate (3x 40 mL). The organic layer was washed with 10% KHSO4, 10% NaHCO3, water and finally brine. The organic layer was dried with Na2SO4, filtered and evaporated to a small volume. The product was purified through column chromatography using silica gel (100-200 mesh) in ethyl acetate/petroleum ether (1:1). Compound 4 was obtained as white solid (80%); melting point: 100 °C; 1H NMR (CDCl3, 300MHz): δ 1.46 (s, 9H, H-1), 3.21 (s, 3H, H-6), 3.72 (s, 3H, H-7), 4.09 (s, 2H, H-4) and 5.28 (brs, 1H, NH); 13C NMR (CDCl3, 75MHz): δ 28.31 (3 x C-1), 32.37 (C-6), 41.68 (C-4), 61.41 (C-7), 79.60 (C-2), 155.86 (C-3) and 170.18 (C-5). IR (KBr) υmax: 3333.33 (N-H str), 3289.71, 3006.83, 2975.75, 2932.75, 1717.43 (COO), 1658.26 (NCO), 1619.57, 1572.61,1447.43, 1401.80,1365.72, 1285.99, 1250.32, 1170.30, 1128.03, 1030.63, 985.00, 946.16, 871.52, 807.40, 780.10, 761.18 and 621.05 cm-1; HRMS: C9H18O4N2 [M]+ 218.2502.
2.5. Synthesis of 2-(bocamino-ethylamino)-3-(4-methoxybenzylsulfanyl)-propionic acid methyl ester (6)
A solution of [(methoxymethylcarbamoyl)-methyl]-carbamic acid tert-butyl ester (3 g, 15.9mmol) in anhydrous diethyl ether (25 mL) was added dropwise in a suspension of LiAlH4 (726 mg, 19.2mmol) in diethyl ether (25 mL) at -30 °C. The reaction mixture was kept at 0 °C overnight and then 10% KHSO4 was added to quench excess of LiALH4. The organic layer was washed with 10% NaHCO3 and brine. The solution was dried on Na2SO4 and the solvent was then evaporated to yield crude aldehyde. The aldehyde (2 g, 12.6 mmol) was dissolved in anhydrous methanol (25 mL) and to it, methyl-2-amino-3-(4-methoxybenzylthio) propanoate hydrochloride (2, 3.7g, 12.6mmol) and NaCNBH3 (1.5g, 25.2mmol) were added. The reaction mixture was stirred overnight at room temperature. Methanol was evaporated and then the residue was dissolved in ethyl acetate. The organic layer was washed with 10% KHSO4,10% NaHCO3 and finally brine. The product was purified through column chromatography using silica gel (100-200 mesh) in ethyl acetate/petroleum ether (1:10). Compound 6 was obtained as viscous oil (40%); 1H NMR (CDCl3, 300MHz): δ 1.45 (s, 9H, H-8), 2.58-2.63 (m, 1H, Hb), 2.67-3.16 (m, 3H, H-4, H-5 and Ha), 3.30-3.39 (m, 1H, H-2), 3.51 (brs, 1H, NH), 3.69 (s, 2H, H-10), 3.73, 3.79 (2 x s, 6H, H-9 and H-11), 5.02 (brs, 1H, NH), 6.85 (d, J =8.4 Hz, 2H, H-2') and 7.23 (d, J =8.4 Hz, 2H, H-3'); 13C NMR (CDCl3, 75 MHz): δ 28.43 (3 x C-8), 34.25 (C-3), 36.18 (C-10), 47.27 (C-4), 52.11 (C-5), 55.29 (C-11), 60.21 (C-2), 60.55 (C-9), 79.17 (C-7), 113.94 (2 x C-2'), 129.83 (C-1'), 130.00 (2 x C-3'),156.04 and 158.72 (C-4' and C-6), 174.03 (C-1). IR (KBr) υmax: 3357.90 (N-H str), 2976.13, 1735.99, 1708.90, 1610.44, 1512.65, 1459.59, 1390.88, 1366.35, 1249.95, 1173.22, 1035.08, 832.51 and 775.30 cm-1. UV (methanol) λmax: 279nm. HRMS: C19H30O5N2S [M]+ 398.8868.
2.6. General procedure for synthesis of 2-[{2-(Base-1-yl)-acetyl}boc-aminoethylamino]-3-(4-methoxybenzyl sulfanyl)-propionic acid methyl ester (7 and 8)
2-(2-Bocamino-ethylamino)-3-(4-methoxybenzylsulfanyl)-propionic acid methyl ester (1g, 2.4 mmol) was dissolved in anhydrous dimethyl formamide (15 mL). At 0 oC, base-1-yl-acetic acid (3.6 mmol) and 1-ethyl-3-(3-dimethylaminopropyl)carbodiimide hydrochloride (EDC) (700 mg, 3.6 mmol) were added. The reaction mixture was stirred overnight and then added to ice cold water (10 mL). The compound was extracted with chloroform and the organic layer was washed with 10% NaHCO3 and brine. The product was obtained through column chromatography using silica gel (100-200 mesh) in ethyl acetate/petroleum ether (3:2).
2.7. 2-[{2-(Thymin-1-yl)-acetyl}boc-aminoethylamino]-3-(4-methoxybenzylsulfanyl)-propionic acid methyl ester (7)
Compound 7 was obtained as white solid (70%); melting point: 68-70 °C; 1H NMR (two rotamers, CDC13, 300 MHz): δ 1.45 (s, 9H, H-9), 1.91 (s, 3H, H-12), 3.09-3.13 (m, 5H, H-6, H-7 and H-2), 3.69-3.79 (m, 10H, H-2', H-4, H-5 and H-15), 4.45-4.61 (m, 2H, Ha and Hb), 5.30 (brs, 1H, NHBoc), 6.85 (m, 3H, H-2'' and H-10), 7.22 (d, J =8.1 Hz, 2H, H-3''); 13C NMR (CDCl3, 75MHz): δ 12.35 (C-12), 28.40 (3 x C-9), 30.53 (C-3), 36.68 (C-4), 38.56 (C-6), 48.13 (C-7), 48.56 (C-2'), 52.85 (C-5), 55.31 (C-15), 61.06 (C-2), 80.00 (C-8), 110.67 (C-11), 114.05(2 x C-2''), 130.02 (2 x C-3'',C-1''), 140.79 (C-13), 150.73, 156.87 and 158.85 (C-4'', C-14 and C-16), 163.98 (C-10), 167.22 and 170.17 (C-1 and C-1'); IR (KBr) υmax 3343.66 (N-H str), 3201.56, 3010.30,2 838.28, 1680.61 (COO), 1610.92, 1512.24, 1465.46, 1368.35, 1302.84, 1247.87, 1174.14, 1107.92, 1032.54, 965.46, 834.78, 755.41 and 667.00 cm-1; UV (methanol) λmax 269nm; HRMS: C26H36O8N4S [M+H+Na]+ 587.9988.
2.8. 2-[{2-(N4-Cbz-cytosin-1-yl)-acetyl}boc-aminoethylamino]-3-(4-methoxybenzylsulfanyl)-propionic acid methyl ester (8)
Compound 8 was obtained as white solid (60%); melting point: 66-68 °C; 1H NMR (two rotamers, CDC13, 300MHz): δ 1.47 (s, 9H, H-10), 3.02-3.38 (m, 5H, H-2, H-6 and H-7), 3.59-3.83 (m, 10H, H-2', H-4, H-11 and H-5), 4.59-4.88, (m, 2H, Ha and Hb), 5.26 (s, 3H, H-17), 5.54 (brs, 1H, NHBoc), 6.88 (d, J=8.1 Hz, 2H, H-2”), 7.26 (d, J=7.8Hz, 2H, H-3”), 7.31-7.51 (m, 7H, Ar-Cbz, H-12 and H-13); 13C NMR (Acetone-d 6 , 75MHz): δ 28.62 (3x C-10), 31.16 (C-3), 36.33 (C-4), 39.82 (C-6), 49.24 (C-7), 50.85 (C-2’), 52.57 (C-11), 55.54 (C-5), 61.49 (C-2), 67.83 (C-17), 79.29 (C-9), 94.67 (C-13), 114.77 (2x C-2”), 128.87, 129.04, 129.36 (C-19, C-20 and C-21), 131.15 (2x C-3”), 132.44 (C-1”), 137.08 (C-18), 151.35 (C-12), 153.91, 156.90, 158.15 and 159.78 (C-4”, C-8, C-15 and C-16), 164.12, 168.39 and 170.90 (C-1, C-1' and C-14); IR (KBr) υmax: 407.37 (N-H str), 3015.79, 2850.36, 1744.75 (COO), 1664.33, 1630.62, 1561.35, 1508.96, 1454.23, 1369.11, 1249.01, 1214.04, 1062.19, 1034.00, 1002.03, 755.25, 698.11 and 667.21cm-1. UV (methanol) λmax: 302nm; HRMS: C33H41O9N5S [M + H]+ 684.1031.
2.9. General procedure for synthesis of 2-[{2-(base-1-yl)-acetyl}boc-aminoethylamino]-3-(4-methoxybenzyl sulfanyl)-propionic acid (9 and 10)
2-[{2-(Base-1-yl)-acetyl}boc-aminoethylamino]-3-(4-methoxybenzylsulfanyl)-propionic acid methyl ester (500 mg) was dissolved in methanol (3mL) and 4M LiOH solution (2 mL) was added to it. The reaction mixture was stirred overnight and then methanol was evaporated. The mixture was acidified to pH 5 by the addition of 2 M HCl solution. The precipitate was then filtered and washed with ether.
2.10. 2-[{2-(Thymin-1-yl)-acetyl}boc-aminoethylamino]-3-(4-methoxybenzylsulfanyl)-propionic acid (9)
Compound 9 was obtained as semisolid (60%); 1H NMR (two rotamers, Acetone-d 6 , 300 MHz): δ 1.42 (s, 9H, H-9), 1.81 (s, 3H, H-12), 3.30-3.50 (m, 5H, H-2, H-6, and H-7), 3.71-3.77 (m, 7H, H-2', H-4 and H-5), 4.29 (brs, 1H, NH), 4.66-4.83 (m, 2H, Ha and Hb), 6.86 (d, J = 8.7 Hz, 2H, H-2”) and 7.25-7.29 (m, 3H, 2 x H-3” and H-10); 13C NMR (Acetone-d 6 , 75 MHz): δ 13.35 (C-12), 29.70 (3 x C-9), 31.24 (C-3), 36.36 (C-4), 39.76 (C-6),49.01 (C-7), 49.19 (C-2’), 55.54 (C-5), 61.39 (C-2), 79.37 (C-8), 109.83 (C-11), 114.78 (2 x C-2”), 114.85 (C-1”), 131.04 (2 x C-3”), 142.62 (C-13), 151.91, 153.46 and 156.86 (C-4”, C-14 and C-15), 159.77 (C-10), 164.86 and 168.40 (C-1 and C-1’); IR (KBr) υmax 3422.33 (N-H and COOH), 2974.78, 2930.22, 1675.98, 1512.18, 1465.01, 1367.77, 1248.77, 1171.39, 1031.70, 964.96, 835.12, 780.41, and 552.03 cm-1; UV (methanol) λmax 268 nm; HRMS: C25H34O8N4S [M+Na]+ 573.4542.
2.11. 2-[{2-(N4-Cbzcytosin-1-yl)-acetyl}Boc-aminoethylamino]-3-(4-methoxybenzylsulfanyl)-propionic acid (10)

Compound 10 was obtained as white semisolid (55%); 1H NMR (two rotamers, CDCl3, 300 MHz): δ 1.43 (s, 9H, H-10), 2.95-3.59 (m, 5H, H-2, H-6 and H-7), 3.64 (s, 2H, H-4), 3.72-3.79 (m, 5H, H-2' and H-5), 4.85-4.96 (m, 2H, Ha and Hb), 5.24 (s, 3H, H-16), 6.21 (brs, 1H, NHBoc), 6.87 (d, J =8.7Hz, 2H, H-2''), 7.28 (d, J = 8.4 Hz, 2H, H-3''), 7.33-7.47 (m, 6H, Ar-Cbz, H-12), 7.87 (d, 1H, J =7.2 Hz, H-11); 13C NMR (CDCl3, 75 MHz): δ 28.43 (3 x C-10), 30.51 (C-3), 36.61 (C-4), 38.82 (C-6), 49.06 (C-7), 49.73 (C-2'), 55.32 (C-5), 61.15 (C-2), 67.89 (C-16), 79.95 (C-9), 94.99 (C-12), 114.02 (2 x C-2''), 128.26-128.69 (C-18 - C-20), 130.10 (2 x C-3''), 131.53 (C-1''), 135.06 (C-17), 149.59 (C-11), 152.35, 155.37 and 155.99 (C-4'', C-8 and C-14), 158.78, 162.91, 167.09 and 170.20 (C-1, C-1', C-13 and C-15); IR (CHC13) υmax 3423.82 (N-H str), 2927.79, 2851.54, 1700.69 (COOH), 1686.74, 1655.94, 1511.48, 1457.34, 1367.83, 1250.34, 1211.18, 1174.83, 1032.25, 833.56, 774.82 and 609.79cm-1; UV (methanol) λmax: 310nm; HRMS: Calculated for C32H37O9N5S [M]+ 667.4619.
3. Results and discussion
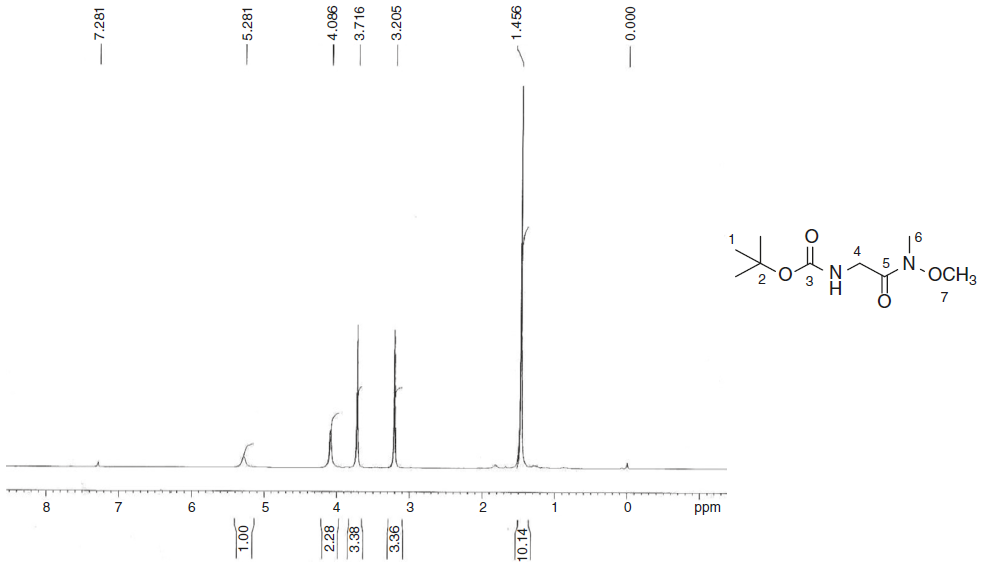
The synthesis of the monomer started with boc-glycine and N,O-dimethylhydroxylamine, which was converted to its Weinreb amide, [(methoxymethylcarbamoyl)-methyl]-carbamic acid tert-butyl ester (4) in about 80% yield. In its IR spectrum, it showed characteristic peaks at 3333 cm-1 that describe N-H stretching. In the 1H NMR spectrum (Fig. 1), a characteristic singlet at δ 1.46 for nine protons and corresponding peaks in 13C NMR at δ 28.31, 79.60 and 155.86 confirm the tert-butyloxycarbonyl (boc) group. Two singlets in 1H NMR at δ 3.21 and δ 3.72 each integrating for three protons were due to methyl and methoxy groups attached to nitrogen, respectively. A singlet corresponding to glycine methylene group was observed at δ 4.09.
Reduction of Weinreb amide with lithium aluminium hydride yields an aldehyde, (2-oxo-ethyl)-carbamic acid tert-butyl ester (5) and its subsequent reductive amination with methyl-2-amino-3-(4-methoxybenzylthio)propanoate hydrochloride (2) yields PNA backbone that contains ligation handle, sulfhydryl group at α-position. Compound (6) was obtained in 40% yield as colourless oil. In its IR spectrum, it showed characteristic peaks at 3357 cm-1 that describe N-H stretching. In the 1H NMR spectrum (Fig. 2), a characteristic singlet at δ 1.45 for nine protons was observed for the three methyl groups of boc and the corresponding carbons in 13C NMR appeared at δ 28.43 while the boc tertiary carbon appeared at δ 79.17. A multiplet in the range δ 2.58-2.63 was assigned to one of the diastereotopic proton Hb. Another multiplet in the range δ 2.67-3.16 was observed for another diastereotopic proton Ha and two methylene groups (H-4 and H-5) of ethylene diamine unit. The corresponding carbons for C-4 and C-5 appeared at δ 47.27 and δ 52.11, respectively in 13C NMR. The chiral centre of cysteine, i.e. H-2 appeared down-field as multiplet in the range δ 3.30-3.39 because of the attached thiol linkage. A singlet for methylene group attached with the sulfhydryl group was observed at δ 3.69, while two singlets corresponding to two methoxy groups were observed at δ 3.73 and δ 3.79 that explain the incorporation of suitably protected cysteine in the backbone at α-position. Two ortho coupled doublets (two protons each) at δ 6.85 (J =8.4Hz) and δ 7.23 (J =8.4Hz) were assigned to the aromatic protons of S-benzyl group. In 13C NMR spectrum, peaks at δ 34.25 and δ 36.18 were identified for two methylene groups, C-3 and C-10 attached to the thiol group. The two methoxy groups were observed at δ 55.29 and δ 60.55, while the carbonyl of the ester group was observed at δ 174.03.
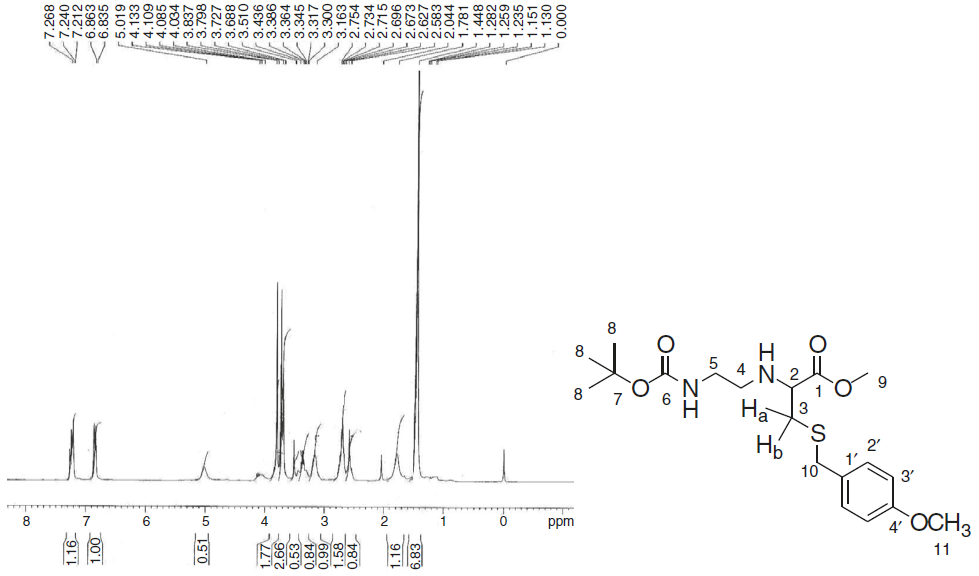
Fig. 2 1H NMR spectra of 2-(Bocamino-ethylamino)-3-(4-methoxybenzylsulfanyl)-propionic acid methyl ester (6).
The thiol group of cysteine was protected using p-methoxy benzyl chloride followed by its esterification in methanol and thionyl chloride to obtain S-p-methoxy benzyl cysteine methylester hydrochloride in about 80% yield (Scheme 1). In its IR spectrum, it showed a characteristic peak at 3412 cm-1 corresponding to N-H stretching. Apeak at 1745 cm-1 explains the presence of ester linkage. In the UV spectrum, it showed absorption at λmax 278 nm. In the 1H NMR spectrum, two multiplets were observed in the range δ 2.88-2.91 and δ 2.98-3.04 for diastereotopic protons Ha and Hb respectively, because of vicinal as well as geminal coupling. A singlet was observed at δ 3.77 for methylene protons of the benzyl group as well as two singlets were observed at δ 3.78 and δ 3.82 which accounts for the presence of two methoxy groups. A downfield triplet for the chiral proton of cysteine (H-3) was observed at δ 4.12 because of the presence of amino and ester functionalities in neighbouring positions. Two doublets at δ 6.88 (J = 8.7 Hz) and δ 7.28 (J = 8.7 Hz) each integrating for two protons was assigned for aromatic protons and this was in accordance with the 1,4-disubstitution of the phenyl group. In the 13C NMR spectrum, characteristic peaks corresponding to carbons of cysteine moiety were observed at δ 31.62 (C-4) and δ 53.27 (C-3). The two methoxy carbons appeared at δ 52.66 and δ 55.97, while the carbonyl carbon was observed at δ 169.52.

The final steps to the target PNA building block comprised the EDC-mediated installation of the suitably protected nucle-obase followed by saponification using 4 M lithium hydroxide in methanol (Scheme 3). Nucleobases thymine and cytosine were incorporated via methylene bridges. Since thymine does not contain any side chain which is to be protected so thymine acetic acid was used as such but the exocyclic amino group of cytosine was protected to prevent self-condensation. The amino group of nucleobase cytosine was protected using the benzyloxycarbonyl (Cbz) group and then introduction of acetic acid was carried out for coupling on backbone (Scheme 2).

In the 1H NMR spectrum for 2-[{2-(N 4 -Cbz-cytosin-1-yl)-acetyl}Boc-aminoethylamino]-3-(4-methoxybenzyl sulfanyl)-propionic acid methyl ester (8) (Fig. 3), a characteristic singlet for nine protons for three methyl groups at δ 1.47 and corresponding peaks in 13C NMR, at δ 28.62 along with δ 79.29 were due to the presence of the boc group. The methylene groups of ethylene diamine backbone (H-6 and H-7) and proton present at the chiral centre of cysteine moiety (H-2) appeared in the range δ 3.02-3.38. The two diastereotopic protons of cysteine Ha and Hb appeared as a multiplet in the range of δ 4.59-k88. The methylene protons of the benzyloxy group (H-17) appeared at δ 5.26 and a multiplet for the aromatic protons of the Cbz group along with the aromatic protons of cytosine (H-12 and H-13) appeared in the range δ 7.31-7.51. That confirms the presence of Cbz-protected cytosine moiety. In the 13C NMR spectrum, peaks for two methylene carbons of ethylene diamine moiety were observed at δ 39.82 and 49.24 for C-6 and C-7, respectively. Characteristic peaks for cytosine were observed at δ 94.67 (C-13) and δ 151.35 (C-12). The amide carbonyl (C-1') and ester carbonyl (C-1) appeared in the range δ 164.12-170.90. In HRMS, the [M+H]•+ peak was observed at 684.1031 having a molecular formula C33H41O9N5S (calculated value: 684.0625).

Fig. 3 1H NMR spectra of 2-[{2-(N 4-Cbz-cytosin-1-yl)-acetyl}Boc-aminoethylamino]-3-(4-methoxybenzyl sulfanyl)-propionic acid methyl ester (8).
In the 1H NMR spectrum for 2-[{2-(Thymin-1-yl)-acetyl}boc-aminoethylamino]-3-(4-methoxybenzylsulfanyl)-propionic acid (9) (Fig. 4), a characteristic singlet for nine protons at δ 1.42 explains the presence of the boc group. A singlet for the methyl group at δ 1.81 for three protons was assigned to the thymine methyl group. The thymine ring proton (H-10) appeared as multiplet along with aromatic protons (H-3”) in the range δ 7.25-7.29. As multiplets in the range δ 3.30-3.50, the methylene groups of ethylene diamine backbone (H-6 and H-7) appear along with the proton present at the chiral centre of cysteine moiety (H-2). In the 13C NMR spectrum (Fig. 5), peaks for two methylene carbons of ethylene diamine moiety were observed at δ 39.76 and 49.01. Characteristic peaks for the methyl group of thymine was observed at δ 13.35. The amide carbonyl (C-1’) and acid carbonyl (C-1) were assigned in the range of δ 164.86-168.40. The identity of the modified PNA monomers was confirmed by NMR, IR and mass spectrometry.

Fig. 4 1H NMR spectra of 2-[{2-(Thymin-1-yl)-acetyl}boc-aminoethylamino]-3-(4-methoxybenzylsulfanyl)-propionic acid (9).

Fig. 5 13C NMR spectra of 2-[{2-(Thymin-1-yl)-acetyl}boc-aminoethylamino]-3-(4-methoxybenzylsulfanyl)-propionic acid (9).
Combining the use of the p-methoxy benzyl group for thiol protection and the Boc group for terminal NH2 has led to the simple preparation of a dual labelled PNA monomer which may be used as a diagnostic tool for biomedical studies. The synthesis of a new mercaptomethyl-modified PNA monomer allows the application of native chemical ligation-like fragment coupling reactions without detriment to backbone geometry. It was shown that the DNA affinity of the PNA ligation products is as high as the affinity of unmodified PNA obtained by linear solid-phase synthesis.
While many PNA backbone modifications have shown the deleterious effect on PNA hybridization, it was observed that the addition of the mercaptomethyl group in PNA ligation products hardly influences the stability of a PNA/RNA duplex. This convergent approach may find applications in the synthesis and labelling of difficult PNA-oligomers and in DNA-template-controlled ligation reactions. The application of suitably protected modified PNA monomers has given access to a PNA oligomer containing a sulfhydryl group appropriate for post-assembly conjugation employing well established conjugation methods.
It has been observed that the incorporation of a sulfhydryl group in ethylene diamine moiety of PNA backbone (γ-exposition) did not substantially affect the hybridization properties with complementary RNA. However, the conjugation of the sulfhydryl group at γ-position leads to the steric destabilization of the PNA-DNA/RNA hybrid. Hence, in this paper we have focused on the synthesis of two thiol-modified α-monomers.
4. Conclusion
Orthogonally protected novel thiol-modified PNA monomers 9 and 10 were prepared containing thymine and cytosine nucleobases. In this report, the thiol modification is carried out in side chain instead of ligation site, i.e. main ethylene diamine backbone. These novel α-monomers would be of relevance in studying hybridization properties and a valuable asset for future conjugation of PNAs with a variety of ligands such as artificial RNA-nucleases for the sequence selective degradation of the target RNA.

