











 nueva página del texto (beta)
nueva página del texto (beta) Inglés (pdf)
Inglés (pdf)
 Artículo en XML
Artículo en XML Referencias del artículo
Referencias del artículo
 Enviar artículo por email
Enviar artículo por email Citado por SciELO
Citado por SciELO  Similares en
SciELO
Similares en
SciELO 
 Permalink
PermalinkIntroduction
Autism (OMIM 209850), also known as autism spectrum disorder (ASD), comprises a complex group of related behavioral disorders with a strong genetic component. ASD is characterized by impairments in social interactions, language, and communication skills, and by restricted and stereotypic behaviors and interests1. Based on its etiology, ASD may be classified as idiopathic, comprising 90-95% of cases; or as secondary, when the autistic phenotype is comorbid to another genetic/non-genetic condition2.
The fragile X syndrome (FXS) is a medical condition strongly associated with ASD. The clinical phenotype includes intellectual and emotional disabilities ranging from learning difficulties to intellectual disability, and mood instability1. In addition, around 30% of children diagnosed with FXS show autism-like neurobehavioral traits, including gaze avoidance, sensory hypersensitivity, social anxiety, stereotypies such as hand flapping and biting, and repetitive language1.
Mutations in the FMRP translational regulator 1 gene (FMR1, OMIM 309550) are responsible for the phenotypic consequences observed in FXS patients1. Most cases are due to expansions of the CGG-repeat number in the 5’-UTR within FMR1 exon 13. Four allelic classes have been defined: (1) normal alleles, (2) intermediate or gray zone alleles, (3) premutation alleles, and (4) full mutation alleles. Normal alleles carry 6-44 CGG repeats. Intermediate or gray zone alleles overlap with normal and premutation alleles, which are normal but slightly unstable (i.e., alleles showing a slightly increased risk of expansion) and could lead to FXS in subsequent generations4. Despite that the American College of Medical Genetics (ACMG) has suggested a range of 45-54 CGGs for these alleles, it has not been accurately defined5-8. Premutation alleles carry 55-200 repeats with an increased risk of expansion to a full mutation through maternal transmission4. Premutation carriers show elevated FMR1 mRNA levels, which have been associated with the development of the fragile X-associated tremor/ataxia syndrome (FXTAS) and fragile X-associated primary ovarian insufficiency4. Finally, individuals with the full mutation carry alleles longer than 200 CGG-repeats, which, in turn, are related to promoter hypermethylation leading to FMR1 silencing and FXS1. The absence of FMRP results in impaired synaptic function and neuroplasticity1.
Although the phenotypic effects of both intermediate and premutation alleles have received considerable attention, the involvement of intermediate alleles in clinical, behavioral, and cognitive phenotypes remains controversial. These alleles have shown a significantly higher frequency in subjects with parkinsonism9,10, ovarian dysfunction11,12, neurodevelopmental delay, and special educational needs13,14, even though some studies have failed to replicate similar results15,16. This study is aimed at determining the distribution of FMR1 alleles in Costa Rican individuals diagnosed with autism, and comparing the allele frequencies of intermediate and full mutation alleles observed in this group with frequencies established for a national birth cohort used as a control group.
Materials and methods
This study was a genetic screening focused on a population of ASD cases from the Central Valley of Costa Rica (CVCR). All cases of suspected autism were collected from this admixed population. A total of 212 participants with autism were recruited: 188 male and 24 female sporadic cases (i.e., only one affected child in each family). Subjects and parents were assessed using the Spanish version of the Autism Diagnostic Interview-Revised17 and the Autism Diagnostic Observation Schedule18. Inclusion criteria, descriptive and clinical data of the probands, ascertainment evaluation process, and clinical testing have been previously published19,20. Parents of participating children provided written informed consent, whereas children also provided signed assent when possible. Blood samples were collected for DNA extraction and genetic FXS testing. The control group consisted of 943 males anonymously enrolled from the Costa Rican National Newborn Screening Program between May and October 2004.
Molecular testing
Genomic DNA was extracted from peripheral blood leukocytes using the proteinase K/phenol-chloroform approach21 and from Guthrie blood spots using the Chelex method22. The number of CGG repeats was determined by polymerase chain reaction (PCR) according to Fu et al.3 with some minor modifications23 using the following primers: FRAXA-A (5´-GTCAGGCGTTCAGCTCCGTTT-3´) and FRAXA-B (5´-CTCCATCTTCTCTTCAGCCCTGCT-3´). PCR was performed in a final volume of 25 ml (100 ng of genomic DNA, 0.4 mM of each primer, 200 mM of each dNTP, 1X DNA polymerase buffer, 15% DMSO, 1.5 mM MgCl2, and 2U of GoTaq DNA polymerase [PROMEGA]). Reactions were carried out in a Perkin Elmer thermocycler GeneAmp PCR System 2400 with an initial denaturation step of 5 min at 95°C, followed by 30 cycles of denaturation at 95°C for 30 s, annealing at 59°C for 1 min, and extension at 72°C for 1 min, with a final 7 min extension step at 72°C. The PCR products were analyzed on 8% polyacrylamide gels and stained with silver nitrate. When PCR was not informative, Southern blot hybridization was carried out. Finally, 5 mg of genomic DNA was digested with Hind III (PROMEGA). The StB12.3 probe for DNA hybridization was used according to the procedure specified by Rousseau et al.24 with some modifications25.
Statistical analyses
Pearson’s χ2 tests were used to compare allele frequencies in the sample of newborns between geographic regions of Costa Rica and to compare frequencies between ASD cases and controls. Data analyses were performed using SPSS software v19 (Chicago, IL). P < 0.05 was considered to be statistically significant.
Results
A total of 1179 X chromosomes were screened for FMR1 CGG repeat number ascertainment, with 212 subjects fitting the diagnostic ASD criteria and 943 newborn males included as a control group. Thirty-two FMR1 alleles were identified in the entire Costa Rican sample (Supplementary Table 1). The normal allele distribution in the control group ranged from 12 to 37, whereas in the case group ranged from 17 to 39 CGG repeats. All the individuals in the control group carried alleles from the normal range. The modal allele was 29 CGGs with a frequency of 37.3% (33.1% in cases and 38.3% in controls) and the second most common allele was 30 CGG-repeats with a frequency of 30.3% (27.1% in cases and 31% in controls) (Supplementary Table 1, Fig. 1).
Table 1 Number of alleles and allele frequency distribution by geographic region in a sample of Costa Rican male newborns (controls)
| CGG-repeats | Central | Caribbean | North | Pacific | Chorotega | All regions |
|---|---|---|---|---|---|---|
| 12-28 | 133 (23.7%) | 34 (30.4%) | 16 (19.1%) | 20 (24.1%) | 24 (23.1%) | 227 (24.1%) |
| 29 | 220 (39.3%) | 38 (33.9%) | 31 (36.9%) | 30 (36.1%) | 43 (41.3%) | 362 (38.4%) |
| 30 | 173 (30.9%) | 31 (27.7%) | 30 (35.7%) | 31 (37.3%) | 28 (26.9%) | 293 (31.1%) |
| 31-37 | 34 (6.1%) | 9 (8.0%) | 7 (8.3%) | 2 (2.4%) | 9 (8.7%) | 61 (6.5%) |
| Total | 560 | 112 | 84 | 83 | 104 | 943 |
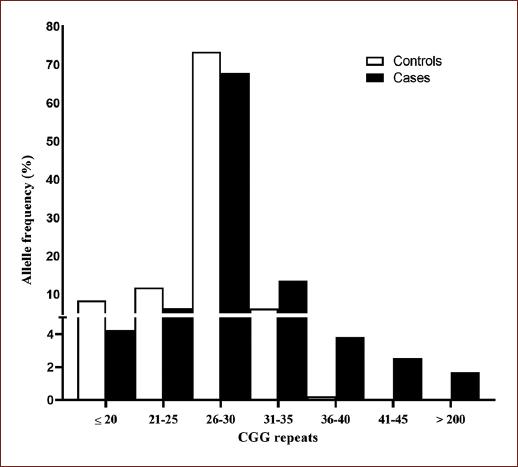
Figure 1 FMR1 CGG-repeat allele size distribution in 943 controls and 212 autism spectrum disorder cases.
Since the newborn control samples were collected from five geographic regions of Costa Rica (Table 1), the allele frequency distribution among these regions was also compared to identify possible geographic effects. However, no significant differences were found between regions (χ2 = 10.41, gl = 12, p = 0.576).
In this study, alleles ranging from 41 to 54 CGG repeats were considered as intermediate or gray zone alleles. Six intermediate alleles (2.5%) in the patient group were found (Table 2). Thus, a significant excess of intermediate alleles was observed in ASD individuals as compared to the control group (χ2 = 24.51, gl = 1, p < 0.001). Furthermore, given that all of the autistic cases were resident from the CVCR region, intermediate allele frequencies observed in this group were compared with frequencies in the control group from the same region. A significant difference between frequencies was found between cases and controls in the CVCR region (χ2 = 14.59, gl = 1, p = 0.001).
Table 2 FMR1 allele frequencies in 943 controls and 212 ASD cases (1179 X chromosomes)
| Allelic form | CGG range | Controls | Cases |
|---|---|---|---|
| Normal | 6-40 | 943 (100%) | 226 (95.7%) |
| Intermediate | 41-54 | 0 | 6 (2.5%) |
| Pre-mutation | 55-200 | 0 | 0 |
| Full mutation | >200 | 0 | 4 (1.7%) |
Finally, no premutation carriers were detected either in the case or in the control group, whereas four full mutation carriers (1.7%) were identified in the patient group (Table 2).
Discussion
This is the first study aimed to investigate FXS prevalence in Costa Rican children and adolescents diagnosed with ASD. The general Costa Rican population as well as the CVCR current population arise from the admixture of ancestral Amerindian, European, and African groups26. As previously reported for populations with West European ancestry and for some Latin American countries, we found that alleles with 29 and 30 CGG-repeats were the more prevalent in the Costa Rican sample27,28. In fact, these alleles accounted for almost 68% of all screened X chromosomes, with the 29 CGG repeat as the modal allele. Such a panorama is consistent with the European ancestral component as the main genetic component for Costa Rican population with a proportion of 49.2%26.
ASD is a neurodevelopmental disorder with an early onset during childhood and is associated with cognitive deficit and behavioral alterations such as a selective impairment in social interaction. Although no accurate tests for identifying the cause of ASD are available thus far, a genetic cause can be determined in some cases. It is widely known that FXS is the most common monogenic cause of autism, resulting in 2-6% of all cases2,29. Here, we found four full expanded alleles in a sample of 212 individuals (236 X chromosomes), representing a frequency of 1.7%. This variation could be explained by differential diagnostic criteria for participants’ inclusion, differences in sample sizes, or population demographic history2. It has been reported that Native American populations have CGG-repeat alleles, and haplotypes related to the polymorphic loci characterized by considerable stability, suggesting a lower prevalence of FXS in these populations30. Taken altogether, these results support the notion that genetic analyses directed to determine FMR1 repeats in ASD subjects are informative in a number of cases, favoring the accurate diagnosis and clinical management for a proportion of cases29,31. DNA testing from blood samples has not been a common practice for the FXS ascertainment in Costa Rican children with ASD, even though an accurate diagnosis is required for specific interventions with particular and appropriate clinical and developmental therapies. For this reason, the screening for FMR1 full mutation alleles is highly encouraged in individuals with idiopathic ASD in Costa Rica and other low- and middle-income countries. Accordingly, given the genetic basis of FXS, additional family members may also take advantage from carrier diagnosis and genetic counseling.
It is worth noting that several discrepancies regarding the definition of the intermediate or gray zone alleles can be found in the literature. Based on the assumption that below 45 CGG-repeats, no mitotic or meiotic instability occurs, the ACMG has established 45 CGGs as the lower and 54 CGG-repeats as the upper boundary7. However, several studies have expanded the range by defining 34-41 CCG repeats as the lower and 60 CGGs as the gray zone upper bound5,8. We considered intermediate alleles as that ranging from 41 to 54 repeats based on molecular findings resulting in altered mRNA expression32,33. Specifically, Loesch et al.33 showed a significant increase in FMR1 mRNA levels in intermediate allele carriers as compared to controls, and a positive relationship between these mRNA levels and CGG-repeat size in carriers within 41-54 repeats, evidencing transcriptional alterations in those individuals.
The frequency of the gray zone alleles was significantly higher in our ASD case group than in the control group, with 2.5% of the individuals carrying an intermediate allele in the case group as compared to 0% in the control group. Remarkably, similar results have been described for 42 Caucasian male children and adolescents34 and for 96 Iranian boys with autism35, suggesting a putative involvement of intermediate alleles in cognitive/behavioral alterations related to autism. This suggestion should be taken cautiously since other studies have failed to replicate this association in cases with ASD or with special educational needs6,8,16,31. Nonetheless, it is worth noting that most of these studies6,8,31 found a non-significant trend toward a higher gray zone allele frequency in cases as compared to controls.
The distribution of intermediate alleles in the general population is quite frequent and ranges from 0.3 to ~4%7 making difficult to draw a conclusion with regard to a relationship between these alleles and autism or other associated phenotypes. Since no robust evidence on intermediate alleles affecting behavioral and/or cognitive phenotypes has been described yet, these findings should be taken cautiously. Additional studies using larger cohorts in different populations with diverse genetic backgrounds should be considered. Furthermore, to establish a possible link between intermediate alleles and clinical outcomes, molecular and functional experiments are also needed.
In that regard, a toxic gain of RNA function has been proposed as a mechanism to explain the increased FMR1 mRNA levels in gray zone allele carriers, and the phenotypic effects documented in some of these individuals. Such mechanism resembles that one observed in pre-mutation carriers affected by FXTAS, where mRNA overexpression leads to intranuclear inclusions in brain cells, aggregation of rCGG-binding proteins involved in RNA processing and translational deficits of FMRP7,32,33. Loesch et al.34 found a positive correlation between the expression levels of FMR1 and the DNA methyltransferase 1 (DNMT1) in intermediate allele carriers, suggesting an alteration of epigenetic mechanisms elicited by elevated FMR1 transcripts. In addition, elevated FMR1 mRNA levels in human cell lines containing premutation CGG-repeat expansions are associated to the upregulation of Ubiquitin Protein Ligase E3A (UBEA3) and Cytoplasmic FMR1 Interacting Protein (CYFIP), which are candidate genes associated with the development of ASD36. In a study with male carriers of intermediate and premutation alleles (40-85 CGG-repeats) affected by parkinsonism, it was found a positive correlation between clinical phenotype (motor and cognitive decline) and CGG-repeat size, antisense FMR1 transcript, and cytochrome C1 mRNAs levels (CYC1) in blood37. Subjects with Parkinsonism carrying intermediate/premutation alleles and with alterations in the CYC1 mRNA levels exhibited significant mtDNA depletion in blood compared with healthy controls and patients with Parkinsonism but with normal FMR1 alleles37. Although this was an unexpected result because CYC1 was initially used as a reference gene, the authors suggested a cytotoxic effect of elevated sense/antisense FMR1 messages that cause mitochondrial dysfunction and correlate with disease severity37. Impairments in mitochondrial function have a high prevalence in ASD subjects as compared to the general population38, and downregulation of CYC1 in postmortem brain tissues from ASD patients has been reported39. As mentioned above, FMRP deficits have also been reported for subjects with elevated FMR1 mRNA levels, therefore, translational dysregulation of other relevant transcripts could also be involved.
Taken altogether, these observations suggest that, unlike the traditional view, intermediate alleles could result in phenotypic alterations potentially relevant for the ASD expression, at least in a minor percentage of patients. Recently, it has been described at the genome-wide level that tandem DNA repeat expansions contribute to the genetic risk of ASD in 2.6%40. A top candidate of tandem repeat-containing regions was selected as ASD relevant in 10 genes including DM1 Protein Kinase, Frataxin, Calcium Voltage-Gated Channel Auxiliary Subunit Beta 1, and FMR1 among others40.
Conclusions
FMR1 screening in subjects with autism should be a common practice since a small fraction of individuals showing the autistic spectrum corresponds to non-diagnosed cases of FXS. We found a significantly increased of intermediate alleles frequency in a sample of Costa Rican children and adolescents diagnosed with ASD compared to a national newborn cohort, suggesting a possible role in the development of this condition. This category of alleles demands more epidemiological studies and a deeper biological comprehension to achieve a more appropriate genetic counseling.

