











 nova página do texto(beta)
nova página do texto(beta) Espanhol (pdf)
Espanhol (pdf)
 Artigo em XML
Artigo em XML Referências do artigo
Referências do artigo
 Enviar este artigo por email
Enviar este artigo por email Citado por SciELO
Citado por SciELO  Similares em
SciELO
Similares em
SciELO 
 Permalink
Permalink1. Introducción
Realizar la reducción de los contaminantes óxido nítrico (NO) y dióxido de nitrógeno (NO2), llamados comúnmente NOx, es una de las tareas más importantes en la química ambiental, debido a que dichos compuestos son causantes de la formación de smog y lluvia ácida. Las principales fuentes emisoras de los compuestos NOx son los automóviles y las industrias, representando el 90% de la producción 1-2. En el caso más sencillo, para llevar a cabo la reducción del NO se utiliza monóxido de carbono para formar nitrógeno y dióxido de carbono:
En la práctica, para realizar esta reacción con eficiencia se utilizan catalizadores bimetálicos de platino-rodio soportados en una mezcla de óxidos de cerio-alúmina (CeO2/Al2O3) 3.
Además del interés ambiental que tiene esta reacción, el estudio de ella también se ha enfocado desde el punto de vista científico. Debido a que al reducir el NO por CO utilizando catalizadores de monocristales de platino y paladio, en condiciones de ultra alto vacío (UHV) se forman fenómenos no lineales, como son: oscilaciones, patrones espaciales y caos 4-6. En particular se ha observado que a p ~ 10-6-10-4 Pa y T > 450 K 5- 7, 8 en el sistema NO+CO/Pt(100) se crean diferentes tipos de fenómenos temporales, entre los que se encuentran: oscilaciones regulares, irregulares, amortiguadas y caóticas 8, así como también una gran gama de comportamientos espaciales, por ejemplo: ondas paralelas, burbujas o células, espirales y turbulencias 4-10. A estas condiciones de presión y temperatura se ha observado que una transición de fase de la superficie (STP) del tipo hex ⇆ 1x1 está relacionada con la aparición de esa variedad de fenómenos espacio-temporales 5-10.
El mecanismo clásico que se ha empleado para explicar la formación de oscilaciones y patrones espaciales en la reacción CO + NO sobre Pt(100) procede de acuerdo a un mecanismo de tipo Langmuir-Hinshelwood (LH), el cual consiste de los siguientes pasos de reacción 5,6,9:
En este esquema de reacción, se puede observar que la producción de N2(gas), se lleva a cabo por la recombinación de 2 átomos de N(ads) que se encuentran en sitios reactivos (átomos de Pt) cercanos, esto es, a primeros vecinos (nn) sobre la superficie de Pt(100).
Por otra parte, el interés en estudiar y explicar la formación de oscilaciones y patrones espaciales en el sistema CO+NO/Pt(100) no sólo ha sido desde el punto de vista experimental, sino que también teórico. Una gran variedad de modelos se han desarrollado al respecto, en los cuales se han utilizado ecuaciones cinéticas derivadas de aproximaciones de campo medio (MF) 5, autómatas celulares (CA) 11 y métodos de Monte Carlo (MC) 6,12, 13, 14.
En la mayoría de estos trabajos se asume que los procesos fisicoquímicos, como adsorción, disociación, difusión, etc., se llevan a cabo sobre un substrato perfectamente periódico, es decir, no se toman en cuenta impurezas y defectos superficiales. Sin embargo, en un sistema real es imposible remover las impurezas completamente por lo que siempre hay una fracción de sitios inactivos que impiden que estos procesos se activen. Por tanto, tales lugares no homogéneos sobre la superficie catalítica pueden afectar la adsorción de las especies desde la fase gas, obstruir los procesos de difusión y de nucleación, por ejemplo 15.
Por estas razones, el presente trabajo tiene como objetivo analizar algunos aspectos del efecto que provocan las impurezas superficiales, marcadas como sitios inertes, principalmente durante la formación de oscilaciones y patrones espaciales, en la reacción de reducción de NO sobre una superficie de Pt(100) utilizando un método de Monte Carlo dinámico. Además se toman en cuenta los parámetros experimentales de cada proceso involucrado en la reacción y se utiliza un mecanismo de tipo Langmuir Hinshelwood.
2. Especificaciones de la superficie de Pt(100)
La superficie limpia de Pt en el plano (100) presenta en su última capa de átomos un arreglo geométrico estable cuasi-hexagonal (hex). Esta estructura puede experimentar una reestructuración hacia un arreglo metaestable cuadrado (1x1) por la adsorción de algún adsorbato como CO o NO, es decir, la estructura adopta la forma geométrica "normal" determinada por el arreglo geométrico del seno del material. Una vez formada la estructura 1x1, las especies adsorbidas pueden ser removidas, por ejemplo por un proceso de reacción, y la superficie regresa de nuevo a la fase hex, es decir se reconstruye a su estructura estable (Figura 1). Este fenómeno forma la transición de fase de la superficie hex ⇆ 1x1, la cual está directamente involucrada con la formación de oscilaciones durante el transcurso de las reacciones de CO+O2 y de NO+CO a temperaturas relativamente altas 5-10.

Figura 1. Reestructuración y reconstrucción de la superficie de Pt(100). Los círculos en azul y blanco indican la primera y la segunda capa de los átomos de Pt, respectivamente.
Las STP inducidas por el adsorbato tienen las bases en simples consideraciones de la termodinámica, en este caso la superficie de Pt(100) se reestructura debido a que la energía superficial de la fase hex es menor a la energía superficial de la fase 1x1, de esta manera la estabilidad de las dos fases cambia si un adsorbato se encuentra con más fuerza enlazado sobre la fase 1x1 que sobre la fase hex, así la energía ganada por la adsorción se compensa con la energía perdida por la reestructuración 5-10,16.
3. Modelo y método de simulación
En el modelo, la superficie del cristal de Pt(100) está representada por una red regular 2D con condiciones de contorno periódicas (cpp). La red consiste de LxL sitios, en donde un sitio indica un átomo reactivo de Pt en la superficie. En la red coexisten ambas fases: hex y 1x1; cada sitio de la fase hexagonal tiene seis sitios a primeros vecinos, mientras que, en la fase 1x1 cada sitio tiene cuatro sitios como primeros vecinos. La fase gaseosa es una mezcla de moléculas de NO y CO con presiones parciales p NO y p CO, respectivamente. En este trabajo se toma en cuenta un esquema alterno de reacción como se indica a continuación:
En donde "sitio" significa un sitio reactivo sobre la superficie. De acuerdo a este mecanismo de reacción los procesos que se han tomado en cuenta en el modelo son:
Adsorción y desorción de CO en las fases hex y 1x1.
Adsorción y desorción de NO en las fases hex y 1x1.
Disociación de NO(ads) en la fase 1x1. Para este proceso se consideran las siguientes características
La probabilidad de disociación de NO(ads) es facilitada por la presencia de átomos de N(ads) que están como nn,
la velocidad de la disociación de NO(ads) aumenta al incrementar el número de sitios vacíos a nn, y
la probabilidad es bloqueada por la existencia de moléculas de CO(ads) y de NO(ads) que se encuentran como nn.
Producción de N2(gas), la cual ocurre por el
paso clásico de recombinación N+N en la fase 1x1 y
por la formación de una especie intermediaria (N-NO)* en la fase 1x1 y la frontera entre ambas fases hex y 1x1.
Además, se considera que el paso (6) es más lento que el paso (5) y, de acuerdo con la referencia 13, ocurren con probabilidades 0.3 y 0.7, respectivamente.
Adicionalmente se han incluido los pasos siguientes (no mostrados en el mecanismo de reacción):
e. Difusión de NO y CO adsorbidos en las fases hex y 1x1 y la frontera entre ellas.
f. Reestructuración de la superficie hex → 1x1.
g. Reconstrucción de la superficie 1x1 → hex.
Para calcular la velocidad de adsorción de las partículas se utiliza la ecuación de Hertz-Knudsen incluyendo las presiones parciales adecuadas. Mientras que las velocidades de los procesos restantes se determinan a partir de la ecuación de Arrhenius utilizando los factores de frecuencia y las energías de activación (cuando sea el caso) de las especies. Los detalles del modelo pueden ser encontrados en las referencias 12 y 13.
Para contar con un modelo relativamente simple y manejable se incorporan las siguientes restricciones:
Los átomos de oxígeno están fuertemente unidos a la superficie del metal y son bastante inmóviles para difundir; de modo que la recombinación de oxígeno atómico y la desorción de oxígeno molecular requieren calentamiento por encima de 600 K 17. Como la temperatura que se va a utilizar en las simulaciones es de 485 K, entonces en el modelo no se toman en cuenta la reacción entre dos átomos de oxígeno y su siguiente desorción como O2(gas).
Un factor que se ignora en el modelo es la diferencia de densidades de átomos de platino que existen entre las fases hex y 1x1, la cual es de aproximadamente 20% más alta en el arreglo hex que en la geometría 1x1 9.
En este estudio el máximo cubrimiento es de un adsorbato por sitio o por átomo de metal, el cual es muy alto. En el sistema real el cubrimiento es menor debido a las interacciones laterales que existen entre los adsorbatos, formándose arreglos c(2x2)-CO(ads) 17 y c(2x4)-NO(ads) 18.
No se toman en cuenta los efectos de los mecanismos de nucleación y crecimiento anisotrópico de las moléculas de CO(ads), aunque éstos han sido observado en la superficie de Pt(100) 19.
Se ignora la adsorción de CO(gas) sobre áreas precubiertas de oxígeno. De los experimentos se conoce que el CO(gas) adsorbe sobre áreas de Pt(100) cubiertas por oxígeno, formando un mecanismo de Eley-Rideal (ER), sin embargo, el mecanismo de ER es mucho más lento que el mecanismo de LH 4.
La producción de N2O(gas) no se toma en cuenta, ya que ésta es muy baja comparada con la correspondiente producción de CO2(gas) y N2(gas 20.
La difusión de los átomos de nitrógeno tampoco se toma en cuenta, en vista de que el N2(gas) es producido también a través del intermediario (N-NO)* y se permite la desorción y difusión de NO(ads). De esta manera, las especies adsorbidas N y NO pueden encontrarse fácilmente para reaccionar. En contraste, cuando se utiliza el esquema de reacción clásico, en donde el N2(gas) es producido sólo por la reacción entre dos átomos de N(ads), se requiere la difusión del N(ads) 20,21.
Debido a que en el modelo no se toman en cuenta la difusión de los átomos de N y O adsorbidos, el paso de reacción (3) no es reversible.
La formación de la especie intermediara (NO-N)* es muy rápida y decae inmediatamente en la formación de nitrógeno molecular. Los experimentos muestran que la producción de N2(g) ocurre rápidamente 17. Por tanto el paso de reacción (4) no es reversible.
Por otra parte, en una simulación dinámica de Monte Carlo (DMC) la evolución temporal del sistema ocurre como un proceso estocástico de tipo Markoviano y la distribución de probabilidad de los estados del sistema está descrita por la ecuación maestra (ME) 22:
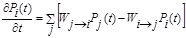 (8)
(8)
donde Pi (t) y Pj (t) son la probabilidades de encontrar el sistema en los estados i y j, respectivamente, al tiempo t. Las W's son las probabilidades de transición por unidad de tiempo para diferentes procesos que se toman en cuenta en el sistema como adsorción, desorción, difusión, etc. Se puede notar que la ecuación (8) simplemente expresa un balance entre el flujo de probabilidades hacia el estado i (ganancia) y el flujo desde i hacia otros estados j (pérdida).
Esta ecuación puede ser resuelta a través de diferentes métodos, para situaciones muy simples, como por ejemplo, para resolver el problema del caminante al azar en 1-D 23. En el caso del sistema considerado en este trabajo, debido a su complejidad que presenta, resolver la ME sería imposible, entonces se puede utilizar una metodología de simulación alterna como la DMC, la cual es viable para reproducir numéricamente la ME. Para ello es imprescindible conocer la relación entre el tiempo físico transcurrido en el proceso y el tiempo de MC o número de pasos de MC realizados. Existen varios métodos que pueden simular la evolución temporal del sistema. Un método que con frecuencia se utiliza en el estudio de reacciones químicas sobre superficies, en el caso particular de la reacción NO + CO, es el "método de selección al azar" (RSM) 13,24. El tratamiento del DMC que se utiliza durante las simulaciones consiste de las siguientes especificaciones:
Un sitio de la superficie se selecciona al azar con una probabilidad 1/N, donde N indica el número total de sitios en la red,
un proceso i se escoge al azar (por ejemplo: adsorción, desorción, difusión, etc.) con una probabilidad Wi/R, donde R es la suma de las velocidades de transición de todos los procesos posibles, es decir, la velocidad de transición total del sistema,
si el proceso seleccionado i puede llevarse a cabo sobre el sitio elegido, entonces ese proceso se efectúa, y
después de que se escoge un sitio, el tiempo se incrementa un Δt, de acuerdo a la ecuación:
 (9)
(9)
donde ξ es un número seleccionada al azar de acuerdo a una probabilidad uniforme en el intervalo (0, 1). Esta ecuación proporciona la evolución del tiempo real causado por una transición del sistema o por un evento fallado.
Para estudiar el efecto de impurezas o sitios inertes sobre el comportamiento oscilatorio y temporal en la reacción de reducción de NO por CO sobre la superficie de Pt(100) se utiliza el algoritmo de simulación siguiente:
Se construye una red de LxL sitios con coordenadas (x,y) y ccp,
se selecciona una fracción de sitios al azar y se marcan como inertes (SI),
se fijan la temperatura del sistema (T) y la presión de los reactantes en la fase gas (Pi ),
se fija el tiempo inicial (t 0 = 0 s),
se determinan las velocidades de los procesos,
se incluye el tratamiento del método de DMC,
se mide el tiempo t de acuerdo a
se acumula t,
se actualiza el tiempo como (t 0 = t), y
el proceso se repite desde el paso 6).
4. Resultados y discusión
Los experimentos indican que la reacción NO+CO en Pt(100) presenta oscilaciones libres, irregulares y caóticas en el intervalo de temperatura de 470-490 K y presiones parciales de las moléculas de NO(gas) y de CO(gas) de ~ 10-4 Pa 8. En el presente trabajo las simulaciones se realizan manteniendo la presión y la temperatura constante del sistema. En el análisis se utilizan las siguientes presiones parciales para los reactantes: p NO = 4x10-4 Pa y p CO = 3.87x10-4 Pa, y una T = 485 K. La Tabla 1 muestra los valores de los parámetros que han sido empleados en las simulaciones, con los cuales se obtienen las velocidades de cada uno de los procesos del sistema. También, en esta tabla se indican los valores experimentales de los parámetros. En este estudio únicamente se toman en cuenta los valores de los procesos en donde se observa la formación de oscilaciones regulares. Cabe señalar que en estudios previos se realizaron simulaciones para este mismo sistema sobre una superficie totalmente limpia y no se incluyó el efecto de sitios inertes. En esos trabajos se analizaron diferentes intervalos para los valores de los procesos involucrados y se hizo un estudio detallado del efecto que tiene cada uno de los procesos durante la aparición de los diferentes fenómenos complejos. De ahí se observó que el sistema presenta una rica variedad de comportamientos oscilatorios y espaciales. Las oscilaciones que se obtuvieron fueron de tipo regular, amortiguadas y moduladas. Mientras que, los patrones espaciales que se observaron consistían en estructuras tipo célula y turbulentos 12,13. Para evitar efectos de borde se utilizan redes de tamaño L = 1024 sitios. Una red de este tamaño garantiza que los frentes de reacción se propaguen y puedan, cuando sea necesario, colisionar entre ellos, incluso por las ccp 12,13.
Tabla 1. Valores de los parámetros utilizados en las simulaciones y la comparación de ellos con los valores observados en los experimentos; en donde p es la presión parcial, S 0 es el coeficiente de adsorción inicial, v es el factor de frecuencia y E a es la energía de activación de las partículas. Los valores experimentales pueden ser consultados en la referencia 13 y las referencias citadas en ese trabajo.

En los resultados del trabajo se observa que los cambios que experimenta la superficie de Pt(100) debido a la STP hex ⇆ 1x1 forman una sincronización entre los reactantes de la fase gas y los procesos fisicoquímicos que ocurren sobre el substrato. Este mecanismo sincronizado causa la aparición de oscilaciones regulares y la formación de patrones espaciales tipo célula sobre la superficie. El mecanismo sincronizado que a continuación se describe es muy similar al encontrado en el sistema CO+O2/Pt(100) 25.
Las simulaciones de la reacción NO+CO sobre el cristal de Pt(100) empiezan con una superficie completamente limpia en el arreglo hex, sobre la cual es posible la adsorción de moléculas de CO y de NO. A un cierto cubrimiento de las especies CO(ads) y NO(ads) sobre la superficie, pequeños núcleos de CO(ads), NO(ads) y mezclas de (CO/NO)(ads) se forman en tal fase y con una probabilidad finita los núcleos pueden transformar algunas pequeñas regiones de la superficie desde la fase hex hacia la fase 1x1, para formar islas de CO(ads), NO(ads) o de (CO/NO)(ads).
Después, esas islas en la fase 1x1 pueden crecer por procesos de entrampamiento: etapa de transformación. El crecimiento de las islas es un proceso estable, pues la reacción puede ser localmente inhibida por un cubrimiento relativamente alto de los adsorbatos, el cual obstruye la disociación del NO(ads). Sin embargo, cuando una molécula de CO(ads) o de NO(ads) desorbe desde la simetría superficial 1x1 hacia la fase gas o cuando una de dichas partículas adsorbidas difunde desde la simetría 1x1 hacia el arreglo hex (los procesos de difusión pueden ocurrir a través de la interfase), siempre y cuando haya desorbido cualquier molécula de la fase hex y su sitio esté como nn, se crea un sitio vacío y si éste tiene una molécula adsorbida de NO a nn, entonces esa molécula de NO(ads) tiene la posibilidad de disociar en un átomo de N(ads) y uno de O(ads). Si el paso de disociación es realizado, entonces el átomo de O(ads) puede reaccionar con una molécula de CO(ads), mientras que el átomo de N(ads) puede también reaccionar, ya sea con una molécula de NO(ads) para formar el intermediario (N-NO)* o con otro átomo de N(ads). Las especies formadas en la superficie pueden desorber como CO2(gas) y N2(gas). Así, nuevos sitios vacíos pueden generarse en la fase 1x1 permitiendo que más moléculas de NO(ads) disocien, por tanto, más átomos de N(ads) y de O(ads) continúan reaccionando, así, al aumentar el número de sitios vacíos en la superficie se permite que el NO(ads) disocie libremente, ocasionando que un frente de reacción empiece a propagarse a través de la superficie: etapa reactiva. Sin embargo, en la fase 1x1 permanecen sitios vacíos y tienen una cierta probabilidad de experimentar la reconstrucción 1x1 → hex, entonces pequeñas áreas de la fase 1x1 pueden lentamente regresar a la configuración hex: etapa de recuperación, por tanto la etapa reactiva se extingue gradualmente. De nuevo, núcleos de CO(ads), NO(ads) y de (CO/NO)(ads) se forman y el ciclo oscilatorio comienza otra vez con la etapa de transformación. En investigaciones experimentales, un ciclo oscilatorio similar fue encontrado para la reacción NO+CO en la superficie de Pt(100), mostrando el papel que juegan los defectos de la superficie, los cuales originan la disociación de NO(ads) en la fase 1x1 8.
El desarrollo anterior es ilustrado en las Figuras 2 y 3. La Figura 2 muestra la formación de oscilaciones regulares obtenidas por simulaciones de DMC en una red de 1024x1024 sitios a una T = 485 K; aquí es posible visualizar la producción de moléculas de CO2(gas) y de N2(gas). Comparando ambas curvas se puede observar que la relación estequiométrica entre el CO2(gas) y el N2(gas) se conserva en una proporción 2:1 como lo indica la ecuación química (1) de la reacción general del sistema. Mientras que la Figura 3 muestra la formación de patrones espaciales tipo célula, los cuales son evidencia de la existencia del mecanismo sincronizado durante el transcurso de la reacción sobre la superficie. Por simplicidad únicamente se muestra la formación de los frentes de reacción (Figura 2(a)) y su crecimiento y propagación a través de la superficie por las ccp (Figura 2(b)).
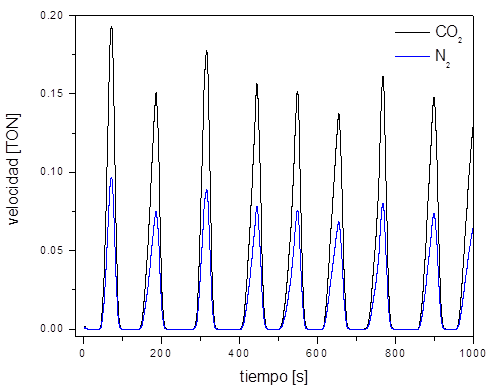
Figura 2. Oscilaciones regulares obtenidas a partir de una superficie limpia. La velocidad de producción de CO2(gas) y N2(gas) está medida en unidades de TON (turn over number, por sus siglas en inglés), es decir, la cantidad de moléculas producidas de CO2(gas) y N2(gas) por átomo de platino por segundo. Las presiones parciales de CO y NO en la fase gas son ≈ 4.0(10-4 Pa, y las constantes de velocidad de los principales procesos son: W desCO = W desCO ≈ 1(10-6 s-1; W difCO = W difCO = 50 s-1; W H→S = 0.015 s-1; W S→H ≈ 0.102 s-1; W disNO ≈ 15.2 s-1

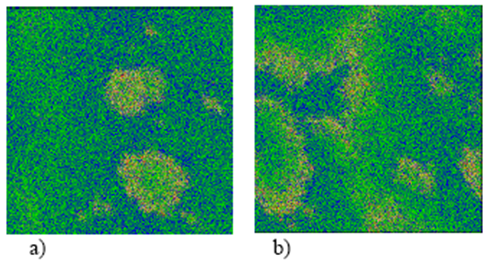
Figura 3. Fotografías correspondientes a la primera oscilación de la Figura 2. Las áreas de color verde indican (CO/NO)(ads) en la fase hex; las áreas de color azul representan (CO/NO)(ads) en la fase 1x1; el color blanco y amarillo indican sitios vacíos en las fases 1x1 y hex, respectivamente, y el color rojo muestra el O(ads). El N(ads) no se alcanza a observar. Los patrones espaciales son tipo célula obtenidos a diferentes tiempos: a) 50 y b) 55 s.
Se puede apreciar que cuando la reacción de NO + CO procede sobre un substrato limpio de Pt(100), es decir no existen impurezas que ocasionen que los sitios de la superficie se desactiven, se tiene un comportamiento oscilatorio y espacial de forma regular, pero ¿qué ocurre cuando el ambiente del substrato cambia? ¿se mantienen estos mismos comportamientos? ¿el comportamiento oscilatorio se mantiene y el espacial desaparece o viceversa? Estas son algunas preguntas que se tratarán de resolver a continuación.
Después de analizar el sistema con una superficie libre de impurezas, se procede a utilizar el algoritmo de simulación explicado en epígrafes anteriores.
Se coloca una pequeña cantidad de impurezas al azar sobre la superficie, ocasionando que los sitios pierdan su actividad, por ejemplo el 3%, es decir, el 3% del total de los sitios de la superficie son inertes. Cuando se bloquea esta cantidad de sitios se observa que el fenómeno oscilatorio cambia drásticamente, esto es, pasa de un régimen regular a uno irregular. En la Figura 4 se puede observar este cambio. Este comportamiento es ocasionado porque la etapa reactiva del mecanismo sincronizado es lenta en comparación con la etapa reactiva cuando se tiene una superficie limpia. Las impurezas ocasionan que las probabilidades para que el CO(ads) reaccione con el O(ads) y el NO(ads) se encuentre con el N(ads) disminuyen. Para que estas especies se encuentren y reaccionen se necesita el proceso de difusión de CO(ads) y NO(ads) el cual está parcialmente bloqueado. En la Figura 5 se puede notar que los frentes de reacción pierden claridad y son menos anchos comparados con los mostrados en la Figura 3, esto es por que la cantidad de oxígeno adsorbido sobre la superficie también ha disminuido, pues hay que recordar que el oxígeno procede de la disociación del NO(ads) y para que este proceso ocurra se necesita un sitio vacío a nn en la fase 1x1, pero la probabilidad para que los sitios cumplan con tal condición ha disminuido. Además, en esta figura también se observa que la cantidad de (CO/NO)(ads) en la fase hex ha aumentado, esto se debe a que el proceso de transformación hex → 1x1 también se encuentra parcialemnte obstruído.

Figura 4. Oscilaciones irregulares obtenidas con 3% de impurezas superficiales. Por simplicidad únicamente se muestra la producción de CO2.

Figura 5. Fotografías correspondientes a la Figura 4 a diferentes tiempos: a) 140 y b) 145 s. Se observa que las áreas de color verde han aumentado, es decir, existe una mayor cantidad de moléculas de CO(ads) y NO(ads) en la fase hex.
Cuando la cantidad de impurezas aumentan al 5% sobre la superficie, las oscilaciones regulares se transforman a un régimen amortiguado. Este hecho se puede observar en la Figura 6. Esto ocurre por tres razones principales: primero se pierde la etapa de transformación, esto ocurre porque la formación de núcleos de CO(ads), NO(ads) y (CO/NO)(ads) en la fase hex disminuyen, provocando también una disminución en la reestructuración de la superficie hacia la fase 1x1; la fase 1x1 se crea principalmente por el proceso de entrampamiento de CO y NO adsorbidos. Segundo se pierde parcialmente la etapa reactiva, el oxígeno tiene poca probabilidad de reaccionar, debido a que la disociación de NO(ads) se encuentra obstruida y los frentes de reacción se extinguen a tal punto que los patrones rompen su simetría, como se puede notar en la Figura 7. Tercero el CO y NO adsorbidos tienen poca probabilidad de difundir, pues los sitios inertes bloquean el salto de éstos a sitios adyacentes. Aunque existe producción de CO2 y N2 ésta se debe a la difusión del CO y NO adsorbidos que logran encontrarse con los átomos de oxígeno y nitrógeno adsorbidos, respectivamente. De esta manera se observa en la Figura 7 que la oscilación se amortigua por que el proceso de reacción no se apaga, pero la cantidad formada de productos disminuye, es decir la sincronía regular entre la fase gas y los procesos que ocurren sobre la superficie cambian para formar otro tipo de patrón oscilatorio (amortiguado) mientras que los patrones espaciales tipo célula se extinguen en su totalidad (Figura 7(b)).

Figura 6. Oscilaciones amortiguadas obtenidas con 5% de impurezas superficiales. Por simplicidad únicamente se muestra la producción de CO2.
En un estudio previo se analizó el efecto de sitios inertes en la reacción de oxidación de CO por O2 utilizando una superficie de Pt(100) 25.
En ese estudio se observó que el comportamiento oscilatorio regular y los patrones tipo célula se pierden cuando existe un 15% de impurezas superficiales mientras que en este trabajo esto ocurre con el 3% de impurezas. Aunado a ello las oscilaciones no pasan por un régimen amortiguado como el encontrado aquí. Comparando estas dos reacciones, se puede analizar que el mecanismo sincronizado de ambas se pierden pero existe una diferencia importante. En la reacción CO + O2 la adsorción del oxígeno es crucial, debido a que el oxígeno molecular solo puede realizar una adsorción disociativa sobre la fase 1x1, es necesario que sobre esta fase existan dos sitios adyacentes como nn para poder realizar este proceso, es decir, la adsorción disociativa del O2 limita los subsecuentes procesos del mecanismo de reacción. Mientras que, en la reacción de CO + NO, el NO puede adsorber en ambas fase y con el apropiado factor de frecuencia y energía de activación puede realizar la disociación en la fase 1x1, es decir, este proceso es independiente del proceso de adsorción, por tanto en este sistema la adsorción de alguno de los reactantes no limita la activación de los otros procesos fisicoquímicos.
Cabe señalar que los resultados reportados en este trabajo están en acuerdo con los estudios realizados por Lui et al.14 al hacer el estudio del efecto de sitios inertes en el mismo sistema. Sin embargo, la diferencia de este trabajo con el de ellos es que se han utilizado diferentes metodologías de Monte Carlo. Ellos utilizaron un Monte Carlo clásico para realizar sus simulaciones y aquí se utiliza un método de Monte Carlo dinámico. Aunque, este trabajo y el de ellos presentan resultados cualitativos similares, en la metodología de Monte Carlo dinámico se tiene la ventaja de que el tiempo real del sistema puede ser medido, es decir, el tiempo "ficticio" de MC clásico puede ser transformado a un tiempo real. Además, en el presente análisis se incluyen los parámetros experimentales de los procesos fisicoquímicos para realizar las simulaciones (ver Tabla 1), mientras que en los trabajos de Lui et al. no se toman en cuenta esos parámetros. Adicionalmente, la diferencia más importantes entre el trabajo de Lui et al. y el presente análisis es que, ellos utilizan el mecanismo clásico de reacción y en este trabajo se utiliza un mecanismo alterno de reacción, en donde se agregan los pasos de reacción (5) y (6), los cuales no han sido descartados a temperaturas altas, mientras que a una T = 450 K se ha podido encontrar experimentalmente el paso de reacción (5) 26. Por tanto, este trabajo de simulación integra características más cercanas a las observadas en los experimentos.
El efecto de los pasos de reacción (4) y (5) son muy importantes en el presente estudio. Por tal motivo, en un estudio previo se hizo un análisis detallado y se encontró que cuando se elimina el paso de reacción clásico (6), N(ads) + N(ads), del mecanismo alterno de reacción, los diferentes fenómenos oscilatorios (oscilaciones regulares, amortiguadas y moduladas) y patrones espaciales (estructuras tipo célula y turbulentos) seguían apareciendo en el sistema. Sin embargo, si la especie intermediaria (N-NO)* se elimina del mecanismo de reacción, en el sistema únicamente se crean oscilaciones amortiguadas y patrones turbulentos 13. Por lo tanto, el paso de reacción (4), formación del complejo activado (N-NO)*, es crucial para obtener los diferentes comportamientos oscilatorios y espaciales en el sistema NO+CO/Pt(100) como están reportados en los experimentos 5,8. Es preciso mencionar que este mecanismo alterno de reacción ha sido observado en el sistema CO+NO/Rh(111) 27, 28, 29 y podría ser válido para otro tipo de superficies, como Rh(100) o Pt(100) en donde la STP hex ⇆ 1x1 está involucrada en la formación de fenómenos espacio-temporales. No existe evidencia experimental (por lo menos a temperaturas altas) que tal mecanismo ocurra sobre la superficie de Pt(100), sin embrago tampoco existen evidencias experimentales directas que digan lo contrario. De esta manera, este y otros trabajos tienen una discusión abierta al respecto 13, 30, 31.
Los argumentos a favor de la existencia del mecanismo alterno de reacción, con respecto a la disociación de NO(ads) y otros procesos que ocurren sobre la superficie de Pt(100), pueden ser reforzados por cálculos cuánticos de la teoría de funcionales de la densidad (DFT) desarrollados por Neurock et al. 32, 33. Ellos muestran que la energía de activación para la disociación de NO(ads) es de 107.1 kJ/mol, mientras que para el proceso inverso es 21.0 kJ/mol. El paso de recombinación N(ads) + N(ads) necesita una energía de 9.0 kJ/mol, la formación de N2O a partir de N y NO adsorbidos necesita 141.9 kJ/mol y para que el N2O decaiga a N2 molecular se necesitan 0.0 kJ/mol. Considerando que dos partículas de NO(ads) deben de disociar para que el paso de reacción N(ads) + N(ads) ocurra, mientras que únicamente se necesita la disociación una partícula de NO(ads) para que el paso de reacción NO(ads) + N(ads) se active y que la energía de activación de la recombinación de NO(ads) a partir de N(ads) y O(ads) es pequeña en comparación a la energía de activación de desorción de NO(ads) (214.3 kJ/mol), la cual aumenta la probabilidad de encontrar partículas adsorbidas de NO sin disociar sobre la superficie, entonces es razonable asumir que ambos pasos de reacción, N(ads) + N(ads) y N(ads) + NO(ads), contribuyen a la formación de N2(gas).
Es pertinente mencionar que aquí no se estudia el efecto de la difusión de las especies adsorbidas, es decir, las velocidades de difusión tanto para el CO(ads) como para el NO(ads) se mantienen constantes, mientras que el estudio reportado Liu et al. estas velocidades cambian. En un próximo estudio se abordará este proceso de manera detallada.
En resumen las impurezas modifican el ambiente del substrato y hacen que el acoplamiento entre la fase gas y éste se pierda, por tanto el mecanismo sincronizado se destruye y provoca la pérdida de los fenómenos oscilatorios regulares y patrones espaciales tipo célula en la reacción de CO + NO sobre una superficie de Pt(100).
5. Conclusiones
En este trabajo se hizo el análisis del efecto que producen las impurezas sobre la superficie de Pt(100) durante la reacción CO + NO utilizando un mecanismo alterno de reacción por simulaciones de Monte Carlo dinámico. Cuando en el sistema no se toman en cuenta las impurezas, es decir, se tiene una superficie totalmente limpia, se puede observar la formación de oscilaciones regulares y la creación de patrones espaciales tipo célula sobre el substrato. Estos fenómenos aparecen debido a que se crea un mecanismo sincronizado entre los reactantes de la fase gas y los procesos fisicoquímicos que ocurren sobre la superficie. Sin embargo, cuando se depositan impurezas o se crean sitios inertes sobre la superficie, la dinámica del sistema se afecta, ocasionando que el comportamiento oscilatorio regular decaiga a un régimen amortiguado y los patrones espaciales tipo célula se destruyen en su totalidad. Este hecho ocurre cuando el 5% del total de los sitios pierden su actividad catalítica. Principalmente, disminuye la creación de núcleos de CO(ads), NO(ads) y (CO/NO)(ads) en la fase hex, por lo tanto la reestructuración de la superficie hacia la fase 1x1 se obstaculiza, la disociación de NO es lenta y el movimiento difusivo del CO y NO adsorbidos se obstruye.

