










Serviços Personalizados
Journal
Artigo
 Inglês (pdf)
Inglês (pdf)
 Artigo em XML
Artigo em XML Referências do artigo
Referências do artigo
 Enviar este artigo por email
Enviar este artigo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO -
 Acessos
Acessos
Links relacionados
-
 Similares em
SciELO
Similares em
SciELO
Compartilhar

 Permalink
PermalinkSuperficies y vacío
versão impressa ISSN 1665-3521
Superf. vacío vol.26 no.2 Ciudad de México Jun. 2013
The modified Becke-Johnson potential analyzed
Camargo-Martínez J.A.1, Baquero R.1
1 Departamento de Física, CINVESTAV-IPN Av. IPN 2508, 07360 México.
Recibido: 11 de diciembre de 2012
Aceptado: 7 de mayo de 2013
Abstract
Recently in the Wien2k code, the modified Becke-Johnson potential (mBJLDA) was implemented. As the authors [Phys.Rev.Lett. 102, 226401 (2009)] point, this potential reproduces the band gap of semiconductors with improved accuracy. In this paper we present our analysis of this potential in two directions. First, we checked whether this potential reproduces the band structure for metals, an analysis that lacked in the literature. We calculated the band gap of a group of semiconductors. We observed that the Linear Density Approximation (LDA) give rise to a shorter lattice constant as compared to experiment. The Generalized Gradient Approximation behaves oppositely. Using the average, aAvg, in the mBJLDA potential, we obtained a closer to experiment value for the gap. We conclude that the new mBJLDA potential represent an important improvement as compared to the results from the previous version of the Wien2k code. Also the mBJLDA potential can be a very useful tool for the theoretical study of complex systems containing semiconductor compounds such as surfaces, superlattices and interfaces.
Keywords: mBJLDA potential; Semiconductors, Band gap; Wien2k 2011.
I. Introduction
Density Functional Theory (DFT) is nowadays the most used method to calculate band structures. It is implemented in several codes. One well known is the Wien2k code. It has evolved in several versions. A friendly interaction was produced already in the 2000 version. Nevertheless, a long standing problem of all the codes based on DFT was that the band structure of semiconductors, in spite of giving a reasonable account of the dispersion of the bands, was systematically unable to reproduce the experimental values of the gap, This problem could be solved by hand using a trick but this kind of solution is not what we expect from an ab initio calculation. The removal of this problem in the new version (Wien2k 2011) is the subject of the analysis that we present in this work. The new ability to reproduce the gap value of semiconducting materials, allows confirming results for semiconductor/metal and semiconductor/semiconductor interfaces and to calculate new ones. These results are of technological interest and per se. For example, the YBCO7/GaAs(001) interface was calculated [1] using the previous version of the Wien2k code (2008 version) and two atomic planes in the GaAs side of the interface were found to be metallic. This result could, nevertheless, be influenced by the inability of the previous version of the code to properly account for the gap of the semiconductor. Since the trick mentioned above could not be used in an interface calculation, the result remains questionable because of the uncertainties in the GaAs side results around the very important gap region. The rest of the paper is organized as follows. In the next section II, we deal very briefly with Density Functional theory (DFT) to point to a detail important to this work. In section III, we briefly present a few of our calculated results for the band structure of metals to check that the accuracy of the new version remains the same also in this case. Section IV is devoted to semiconductors. We recalculate the results from references [2] and [3] and present new results for some other semiconductors which we compare to experiment. In a final section V, we analyze the results with the new code and present our conclusions.
2. Density Functional Theory
In solids, ions and electrons constitute a many body interacting system described by a Schrödinger equation with too many particle coordinates to be numerically treatable nowadays, as it is very well known. During the last decade, several codes were developed based on DFT and this method became the most used, precise and practical way to calculate the band structure of solids. The development of practical approximations to the correlation and interchange potential lead to a remarkable degree of accuracy to describe even complicated metallic systems. At the basis of DFT is the celebrated Hohenberg-Khon theorem which shows that the density of the ground state contains all the possible information on a system and its knowledge is equivalent to the wave function itself. So, the expectation value [4] of any observable can be calculated from a unique functional of the ground state density, ρ(r), which minimizes the energy functional, E[ρ]. Further, Khon and Sham [5] transformed the many-body problem into a one-body problem and showed that the density f states calculated from the solution of the so-called Khon-Sham equations (1) is equal to the one of the real ground state density of the many-body system

where the density is calculated taking into account the occupied states only. In eq. (1), T is the kinetic energy operator, VH is the Hartree potential and Vxc is the exchange and correlation potential which is calculated from the exchange and correlation energy functional, Vxc(r)=δExc[p]/δp. To solve the Khon-Sham equations (1), an explicit expression for Exc[ρ] is needed. The exact expression is unknown since it includes all kind of correlations between all the particles in the system. So an approximation is needed. The first and best known approximation is the Local Density Approximation, LDA [6], which was followed by the Generalized Gradient Approximation (GGA) [6] and the meta-GGA [7] among other approximations. These potentials reproduce rather well the band structure of even complicated metallic systems but fail in reproducing the gap in semiconductors. A recent progress has been made. Blaha et al. [2] have reported the so-called mB-JLDA potential which is a modification of the exchange and correlation potential of Becker and Johnson (BJ) [8]. The new potential reproduces the experimental gaps of semiconductors with accuracy of several orders of magnitude better than the former existing potentials. The modified mBJLDA potential is
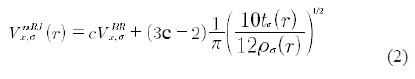
where ρσ(r) is the density of states, tσ is the kinetic energy density and VxσBR (r) is the Becke-Roussel potential (BR) [9]. The c stands for,
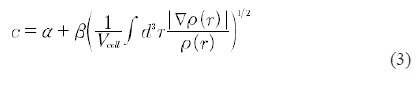
α and β are free parameters. Within the Wien2k code [10] α = -0.012 and β = 1.023 Bohr1/2.
3. Metal Calculations
We present here our result for Nb, V and Ta, to check whether differences arise between the LDA and the new mBJLDA potential for the case of metal. This analysis does not appear reported in the literature, and are important to the calculation of metal/semiconductor interfaces. We have calculated these band structures, first, using the LDA approximation with the old Wien2k code and then we redid the same calculation using the new mBJLDA potential. To optimize the lattice parameter in a consistent way it is recommended [2] to use LDA (GGA) first and to use further the optimized lattice parameter obtained in this way to compute the band gap structure with the mBJLDA potential. If we follow this method, we find a good agreement between LDA and mBJLDA for all the three metals calculated as it can be checked from Table I.

The Fermi energy values, EF, calculated with the mBJLDA potential, for Nb, differs in 0.01 Ry with respect to the LDA value, which represent a difference of 1.3%. For V and Ta these values are 0.2% and 1.8% respectively. The density of states at EF, N(EF), presents difference of 7.7%, 3.4% and 2.0% for Nb, V and Ta respectively. We omit the plot of the band structure and the density of states obtained in the different ways mentioned here since the overall agreement is such that the details just discussed do not show explicitly enough and these band structures are very well known. For Nb we have compared our results with references [12-14], V with references [14, 15] and for Ta with reference [16]. These results show that the mBJLDA potential reproduces well the band structure of metals.
4. Semiconductor Calculations
A particular feature of mBJLDA potential is that a corresponding exchange and correlation energy term , Exc[ρ], such that the mBJLDA potential is obtained in the usual way, namely, Vxc = δExc[ρ]/δρ, is not possible. As a consequence, a consistent optimization procedure to obtain the lattice parameter, the Bulk modulus and its derivative with respect to pressure are not actually possible. This is a consequence of the empirical character of this potential. For that reason, Tran and Blaha have proposed the empirical alternative that prior to a band structure calculation with the mBJLDA potential, the lattice parameter is found from either a LDA or a GGA optimization procedure and the result introduced into the code to perform the band structure calculation of the semiconductor system. Such a procedure gives rise to quite improved results as compared to the previous version of the Wien2k code, as we stated before. It is known that the LDA underestimates as a rule, the lattice parameters and, on the contrary, GGA overestimates them. We have explored the possibility of using the averaged value as the lattice parameter, aAvg, where aAvg = (aLDA + aGGA)/2. Here aLDA(aGGA) is the lattice parameter obtained from an LDA (GGA) optimization procedure. When aAvg is used as input into the Wien2k code implemented with the mBJLDA potential, a better agreement of the band gap value with experiment is obtained as compared to the results with aLDA. So this procedure turns out to give better results than the one recommended by Tran and Blaha and its extra computational cost is relatively low.
In Table II, we present the gap value obtained from our band structure calculations for several semiconductors using LDA and the new mBJLDA potential, using as lattice parameters aLDA and aAvg, called mBJLDA(aLDA) and mBJLDA(aAvg) respectively, and compare our results with the ones reported by Blaha et al. [2], with the ones obtained using the hybridized exchange potential of Heyd-Scuseria-Ernzerhof (HSE) reported in reference [3] and to the experimental values reported in references [2, 3, 17, 18].

It is evident from Table II that the gap values for semiconducting systems calculated with LDA turn out to be wrong as it is very well known. We can see clearly that the results obtained using the mBJLDA potential shows a significant improvement in the calculation of the gap with respect to experiment. Now, the values calculated with mBJLDA(αAvg) have a significantly better agreement with experiment of the gap as compared to the one obtained with mBJLDA(αLDA) and with the values reported by Blaha. The average absolute error values for each procedure were 8.2% for mBJLDA(αAvg), 9.3% for values reported by Blaha and 10.8% for mBJLDA(αLDA). The results calculated with the HSE (See Table II) present a good agreement with experiment too. We conclude that our proposal to use the average value, αAvg, for calculating the band gap using the mBJLDA potential in the Wien2k code results in better agreement with the experimental values. The new mBJLDA potential opens the possibility to carry out theoretical studies of complex systems containing semiconductor compounds as surfaces, superlattices and interfaces.
5. Conclusions
In this work we performed an analysis of the new progress done in the implementation of DFT whose most famous shortcome was its impossibility to account for the experimental value of the band gap of semiconducting systems. We have calculated using the new mBJLDA potential [2], the band structure of some semiconductors and got their band gap value which we compared to experiment. In this work, we found two important facts. First, the mBJLDA potential reproduces correctly the band structure of metals, which is an important new observation. This result is not reported in the literature. Second, that the best result for the band gap value is obtained, in general, if the average lattice parameter, αAvg, is used (αAvg = (αLDA + αGGA)/2) where αLDA(αGGA) is the lattice parameter that results from a LDA(GGA) optimization, We have calculated the band gap for all semiconductors reported in ref. [2] and ref. [3] and some other and compare the results among themselves and with experiment. We found that the new mBJLDA potential gives rise to gap values that represent an important progress as compare to the old LDA potential. This new mBJLDA potential, allows the calculation of interfaces and superlattices with a semiconducting component with a high degree of accuracy which were diffcult with the old code. For example, in the ref. [1] the electronic band structure of the interface YBCO7/GaAs was calculated using Wien2k 2008 code and obtained that two atomic planes in the GaAs side become metallic. Nevertheless since the code does not allow the correct calculation of the gap of the semiconductor, this interesting result remained uncertain [19]. In that sense, the new potential opens a new field which is of interest in several disciplines as spintronics, semiconductor devices, superconductivity or two-dimensional electron gas properties, among others.
Acknowledgments
The authors acknowledge to the GENERAL COORDINATION OF INFORMATION AND COMMUNICATIONS TECHNOLOGIES (CGSTIC) at CINVESTAV for providing HPC resources on the Hybrid Cluster Supercomputer "Xiuhcoatl", that have contributed to the research results reported within this paper.
References
[1]. A. E. Garcia, J. Camas. S. Mendoza, R. Baquero-Salaguarda, L. M. Garcia-Cruz, R. Baquero, The Icfa J. Phys. I, No 4, (2008). [ Links ]
[2]. F. Tran, P. Blaha, Phys. Rev. Lett., 102, 226401, (2009). [ Links ]
[3]. J. Heyd, J.E. Peralta, G.E. Scuseria, R.L. Martin, J. Chem. Phys., 123, 174101, (2005). [ Links ]
[4]. P. Hohenberg, W. Khon, Phys. Rev., B 136, 13864, (1964). [ Links ]
[5]. W. Khon, L.J. Sham, Phys. Rev., 140, A1133, (1965). [ Links ]
[6]. J.P. Perdew, Y. Wang, Phys. Rev., B 45, 13244, (1992). [ Links ]
[7]. J.P. Perdew, S. Kurth, J. Zupan, P. Blaha, Phys. Rev. Lett. 82, 2544, (2000). [ Links ]
[8]. A.D. Becke, E.R. Johnson, J. Chem., 124, 221101, (2006). [ Links ]
[9]. A. D. Becke, M.R. Roussel, Phys. Rev., A 39, 3761, (1989). [ Links ]
[10]. P. Blaha, K. Schwars, G.K.H. Madsen, D. Kvasnicka, J. Luitz, WIEN2K:Full Potential Linearized Augmented Plane aves and Local Orbital Programs for Calculating Crystal Properties, edited by K. Schwars, Vienna University of Technology, Austria, (2001). [ Links ]
[11]. K. Wang, R.R. Reeber, Mat. Sci. Eng., R23, 101, (1998). [ Links ]
[12]. B. Koslowski, C. Dietrich, P. Ziemenn, Se. Sci, 557, 225, (2004). [ Links ]
[13]. A.R. Jani, N.E. Brener, J. Calaway, Phys. Rev., B 38, 9425, (1988). [ Links ]
[14]. O. Madelung, Data in Sciene and Technology, Landolt-Borstain Group III: Crystal and Solid StatePhysics, Vol 13 Metals; Phonon States, Electron States and Fermi Surfaces, Subvolume C. Ed. Springer-Verlag, (1984). [ Links ]
[15]. D. Papaconstantopoulos, J. Anderson, J. McCaffrey, Phys. Rev., B 5, 1214, (1972). [ Links ]
[16]. I. Petroff, C. R. Viswanathan, Phys. Rev., B 4, 799, (1971). [ Links ]
[17]. S. Ada Chi, Handbook on Physical Properties of Semiconductors, Vols. I, II, III. Kluwer Academic Publishers, (2004). [ Links ]
[18]. L. C. O. Dacal and A. Cantarero, Solid State Comm., 151, 781, (2011). [ Links ]
[19]. Preliminary calculations with Wien 2011 seem, nevertheless, to confirm the result obtained previously.

