










Servicios Personalizados
Revista
Articulo
 Español (pdf)
Español (pdf)
 Artículo en XML
Artículo en XML Referencias del artículo
Referencias del artículo
 Enviar artículo por email
Enviar artículo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO -
 Accesos
Accesos
Links relacionados
-
 Similares en
SciELO
Similares en
SciELO
Compartir

 Permalink
PermalinkSuperficies y vacío
versión impresa ISSN 1665-3521
Superf. vacío vol.26 no.2 Ciudad de México jun. 2013
Crecimiento y caracterización de nanofibras obtenidas a partir de proteína aislada de suero lácteo y oxido de polietileno por la técnica de electrohilado
Colín-Orozco J.1, Zapata-Torres M.1, Pedroza-Islas R.*2, Rodríguez-Gattorno G.3
1 Centro de Investigación en Ciencia Aplicada y Tecnología Avanzada del Instituto Politécnico Nacional, Legaría 694. Colonia Irrigación, 11500 México D. F.
2 Universidad Iberoamericana. Prolongación Paseo de la Reforma 880. Lomas de Santa Fe, D.F. * ruth.pedroza@ibero.mx
3 Departamento de Fisica Aplicada, CINVESTAV-IPN, Unidad Mérida, Mérida Yucatán, 97310, México.
Recibido: 4 de enero de 2013
Aceptado: 15 de abril de 2013
Resumen
En el presente trabajo se obtuvieron nanofibras de proteína aislada de suero de leche (WPI por sus siglas en ingles) y óxido de polietileno (PEO por sus siglas en ingles) empleando la técnica de electrohilado. Para la preparación de las fibras se parte de dispersiones de ambos materiales variando la proporción de PEOxWPI100x, en el rango de 0≤x≤100. Las nanofibras obtenidas fueron caracterizadas por Microscopía Electrónico de Barrido, Difracción de Rayos-X y Termogravimetría. A valores de x entre el 100% y 40% de contenido de PEO se favorece el crecimiento de fibras con diámetros promedio que oscilan alrededor de los 230 ± 80 nm, mientras que las dispersiones con alto contenido de WPI promueven la formación de morfologías tipo "perlas". La cristalinidad de las nanofibras incremento a medida que aumenta la cantidad de PEO en la solución. Las nanofibras obtenidas de las combinaciones PEO60WPI40 y PEO50WPI50 fueron térmicamente más estables.
Palabras clave: Nanofibras; Electrohilado; Óxido de polietileno; Proteína aislada de Suero Lácteo.
Abstract
In this work were obtained nanofibers of whey protein isolated (WPI) and polyethylene oxide (PEO) using the electrospinning technique. For the preparation of the fibers we started of dispersions of both materials by varying the proportion of PEOxWPI100-x, in the range of 0≤x≤100. The nanofibers obtained were characterized by scanning electron microscopy, X-ray diffraction and theermogravimetric analysis. For values of x between 100% and 40% of PEO content, The fibers growth with diameters average around the 230 ± 80 nm, whereas the dispersions with high content of WPI promote the formation of morphologies type "pearls". The crystallinity of the nanofibers increased with the increases of PEO concentration in the solution. The nanofibers obtained the combinations of PEO60WPI40 y PEO50WPI50were more thermally stable.
Keywords: Nanofibers; Electrospinning; Polyethylene oxide; isolated whey protein.
1. Introducción
El electrohilado, es un proceso para formar fibras de diámetro nanométrico a micrométrico, cuyo principio básico consiste en aplicar un alto voltaje (entre 5 kV y 30 kV) a una solución polimérica la cual, al cargarse eléctricamente, se deforma en hilos que se entrelazan al dirigirse hacia una zona de más bajo potencial, la cual es generalmente conectada a tierra [1,2]. El control sobre los parámetros de preparación permite disponer de fibras que presentan características muy deseables en materiales compuestos como son: alta área superficial específica, alta porosidad y tamaño de poros muy pequeño [3]. Este proceso es comúnmente aplicable a diversos tipos de polímeros [3] y recientemente se han elaborado nanofibras de biopolímeros con base en proteínas y polisacáridos, solos o combinados tales como la zeína [4], proteínas de trigo [5], alginato de sodio/pectina [6], gelatina/alginato de sodio [7] y zeína/chitosan [8]. No obstante, la literatura sobre la fabricación de nanofibras de biopolímeros es limitada, dado que su producción es usualmente más difícil en relación a la de polímeros sintéticos [9].
La preparación de nanofibras de polímeros naturales, de manera general incluye la incorporación de algún polímero sintético como el óxido de polietileno (PEO) para mejorar la hilabilidad. Se ha reportado el crecimiento de nanofibras utilizando la combinación del PEO con polímeros naturales; por ejemplo albúmina de huevo/PEO [10], alginato de sodio/ PEO [11,12], keratina/PEO [13] y recientemente de mezclas de WPI100 xPEOx, estudiando el efecto del pH para valores de x de 10% w/w y 0.4% w/w [14].
El PEO es un polímero biodegradable y biocompatible, soluble en agua, capaz de formar puentes de hidrógeno con otros polímeros, además de mostrar propiedades como de retención de agua, floculante, espesante, lubricante, dispersante y de liberación sostenida; es no iónico, no tóxico, no irritante y no genera residuos, sedimentos o elementos gaseosos [15,16]. Además, se encuentra disponible en diferentes pesos moleculares y tiene diversas aplicaciones en la industria farmacéutica para la preparación de hidrogeles, liposomas, nano partículas y micropartículas como sistemas de liberación de ingredientes activos [17,18].
Por otra parte, la proteína aislada de suero de leche es una proteína de gran interés debido a sus excelentes propiedades funcionales, alto contenido en aminoácidos esenciales y disponibilidad [19,20]. El WPI tiene buenas propiedades de actividad superficial y ha sido ampliamente utilizado en la industria alimentaria como emulsificante, estabilizador, gelificante y agente espumante, blanqueador o potenciador del sabor [21,22]. Diversas investigaciones han utilizado WPI para producir emulsiones estables así como películas transparentes [23], flexibles, incoloras e inodoras las que han mostrado ser excelentes barreras al oxígeno [24] y humedad [20], inhibiendo la degradación de los componentes funcionales inestables que protegen [22].
La ciencia de los alimentos ha mostrado interés específico en la producción de nanofibras a partir de polímeros biodegradables y biocompatibles, como respuesta a la necesidad de desarrollar materiales de empaque con mejores propiedades mecánicas y barrera y por la demanda de un mejor desempeño de los sistemas de micro encapsulación [10]. Por otra parte, las nanofibras, por sus dimensiones, ofrecen el potencial de mejorar significativamente la solubilidad y la biodisponibilidad de muchos ingredientes funcionales, así como el comportamiento de la liberación controlada [25]. En este trabajo se estudió la influencia de la variación de las proporciones PEO/WPI sobre la morfología de las nanofibras obtenidas por la técnica de electrohilado, así como sus propiedades estructurales y térmicas.
2. Materiales y Métodos
La proteína aislada de suero de leche (WPI) con 90% de proteína fue obtenida de Davisco Foods International, Inc. (EUA). Los datos proporcionados por el fabricante para el WPI son: proteína 97.6 %, ceniza 2%, Grasa < 0.5%, lactosa 0.4 %. El óxido de polietileno (PEO, 300 kDa), fue adquirido de Sigma-Aldrich (EUA). Como solvente se utilizó agua bidestilada, producida por un sistema de filtración Milli-Q. Se prepararon dispersiones de PEO y de WPI con concentraciones de 9 p/p% 10 p/p% , respectivamente; estas dispersiones se dejaron en refrigeración durante 12 h para su completa hidratación. La dispersión de WPI fue desnaturalizada a una temperatura de 85°C por 30 min. Se prepararon mezclas de las dispersiones poliméricas, variando la concentración de ambos polímeros para tener las siguientes proporciones en peso PEO/WPI: 100:0, 80:20, 70:30, 60:40, 50:50, 40:60, 30:70, 20:80 y 0:100. Las mezclas fueron mantenidas a temperatura ambiente (20 °C) en agitación constante durante 3 h previo al electrohilado.
El equipo de electrohilado utilizado se esquematiza en la figura 1. Consiste en una fuente de alto voltaje (Modelo 210-30P, Bertan Assoc Inc., USA), una bomba de infusión para controlar la tasa de flujo de inyección (Modelo NE-300. New Era Pump Systems, Inc., USA), una jeringa de 5 mL con una aguja de acero inoxidable con un diámetro interno de 4.7 mm (25G) (Terumo Corporation, Philippines) y un conector a tierra. El equipo se colocó en una cámara de acrílico para reducir el efecto de las corrientes de aire sobre la trayectoria del flujo electro-depositado. La solución polimérica fue cargada en la jeringa y en la punta de acero inoxidable se aplicó un voltaje de 20 kV. La velocidad de flujo fue de 0.05 mL/h y la distancia de la punta de la aguja al colector de 20 cm.
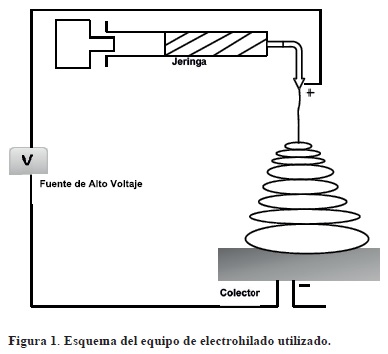
La morfología de las nanofibras fue observada en un microscopio electrónico de barrido (SEM) (JEOL JSM-6390LV, Akishima, Japón). El colector de aluminio con fibras depositadas fue adherido al porta muestra con cinta carbón doble adhesiva y fue cubierto con Au/Pd durante 2 min en un evaporador Desk IV CTC Parker Modelo Standard (Denton Vacuum LLC. NJ EUA). La distribución de diámetros de las fibras fue determinado por análisis de imágenes (Scandium, Universal SEM Imaging platform, Image, JEOL, EUA) a partir de 100 fibras/muestra.
La estructura cristalina de las muestras se determinó utilizando un difractómetro de rayos-X (BRUCKER D8, Alemania). Los difractogramas se adquirieron en el rango de 10° a 60° en 2θ con un paso de 0.02°.
Se determinó el cambio de peso de las fibras en función de la temperatura usando un analizador termogravimétrico Modulado de Alta Resolución (TGA Q 5000, TA Instruments, Inc., USA).
Las muestras se colocaron en la balanza del sistema (0.5-1μg) y fueron calentadas de 25°C a 700°C en porta muestra de platino (platinum-HT) a una velocidad de calentamiento de 7°C/min con una atmósfera de nitrógeno con una velocidad de flujo de 50 mL/min [10].
3. Resultados y Discusión
En la ausencia de PEO, la solución biopolimérica no pudo ser electrohilada. En la figura 2 se presentan las micrografias obtenidas en las mediciones en el SEM, así como un acercamiento a menor escala para una mejor apreciación. En la figura 3 presentamos la variación del diámetro de las fibras obtenidas de las soluciones con diferentes combinaciones de PEO y WPI, en esta grafica se consideró la media aritmetica y como barra de error una desviación estándarde las mediciones, en la cual puede observarse que no hay una dependencia del diámetro con la composición para valores mayores al 40 % en PEO. Las fibras obtenidas de las mezclas PEO100WPI0, PEO80WPI20 y PEO70WPI30 tienen un promedio de diámetro de 243±47.03, 227±36.32 y 264±66.11 nm, respectivamente, mientras que las fibras obtenidas de la mezcla PEO70WPI30 presentaron un promedio de diámetro mayor (264±66.11 nm). A partir de la combinación PEO60WPI40, PEO50WPI50 y PEO40WPI60 hubo una disminución lineal del tamaño de diámetro, 232±35.29, 230±63.10 y 227±67 nm, respectivamente.


La desnaturalización de la proteína promovió la formación de las fibras debido a los enlaces intra e intermoleculares de los grupos amino, carboxílicos e hidroxilo que contribuyen a la estructura α-hélice de la proteína, además de la formación de puentes de hidrógeno con las moléculas de PEO [26]. Al mismo tiempo, se favorece la ionización y por tanto la conductividad aumenta, lo que se ve reflejado en el nivel de elongación del jet durante el proceso de electrospinning; no se produjo un exceso de cargas superficiales en la solución que impidiera el estiramiento de la fibra, al contrario de lo que mencionan Vega-Lugo y Lim [14], ya que el PEO es un polímero neutro y las mezclas de PEO y WPI estaban en un ambiente de pH de neutro a ligeramente alcalino (Tabla 1).

No obstante, las fibras obtenidas con las soluciones PEO30WPI70 y PEO20WPI80 presentaron "perlas" en su morfología con un promedio de diámetro de 648±121.20 nm y 561±110.18 nm, respectivamente (figuras 2g y 2h). Estos defectos pudieron deberse a que la solución no pudo ser completamente estirada durante la electroeyección, lo cual pudiera deberse a la baja viscosidad de sus dispersiones, ya que también se ha reportado que a baja viscosidad generalmente las interacciones entre las cadenas de los polímeros se reducen, incrementando la probabilidad de que se formen "perlas".
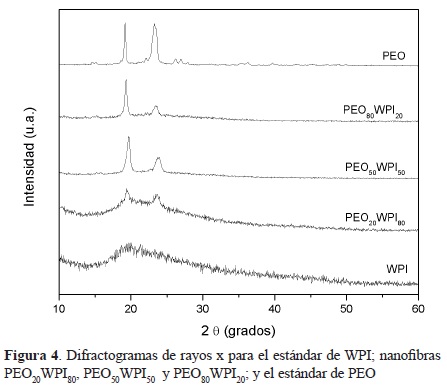
La Fig. 3 muestra los dfractogramas de rayos X de las nanofibras obtenidas con las soluciones PEO20WPI80, PEO50WPI50 y PEO80WPI20; así como los obtenidos a los estándares de WPI y PEO. Se puede notar que el estándar de WPI presenta un espectro característico de una estructura amorfa, mientras que el estándar del PEO presenta picos bien resueltos en el rango de 15° a 30°. En los difractogramas de las nanofibras se pueden observar picos intensos a 19.30° y 23.50°. El pico situado en 2θ=19.30° está relacionado con el plano cristalográfico (120); y el pico en 2θ=23.50° puede corresponder a varios planos: (032), (132), (112), (212), (004) y (124) [27]. Es de hacer notar que a medida que se va incrementando la cantidad de PEO en las nanofibras, va aumentando la cristalinidad; esto se evidencia por el incremento en la intensidad en los picos, así como en la pérdida del halo amorfo correspondiente al WPI.
Las curvas termogravimétricas del porcentaje de peso de las nanofibras, con respecto al peso inicial en la medición (W), de las diferentes mezclas de PEOxWPI100-x y de los materiales solos (PEO y WPI) en función de la temperatura, se muestran en la figura 5. Se observa que el PEO puro en polvo se descompone en una sola etapa en un rango de temperaturas de 300 °C a 406 °C, con una velocidad de descomposición máxima a la temperatura de 380°C y una pérdida de peso de 97% (Figura 5a). En las fibras obtenidas de la solución de PEO100WPI0 se encontró una temperatura máxima de degradación de 362°C, esto es 18°C menor que el PEO puro en polvo, presentando una pérdida de peso de 98%. El WPI empieza con una rápida pérdida de peso inicial de alrededor del 7%, probablemente asociada a un proceso de deshidratación de agua fisisorbida y alcanza una temperatura máxima de degradación a los 292°C, y va en un rango de 250°C -330°C con un 68.26% de pérdida de peso, producto de la descomposición (Figura 5a y 5b).
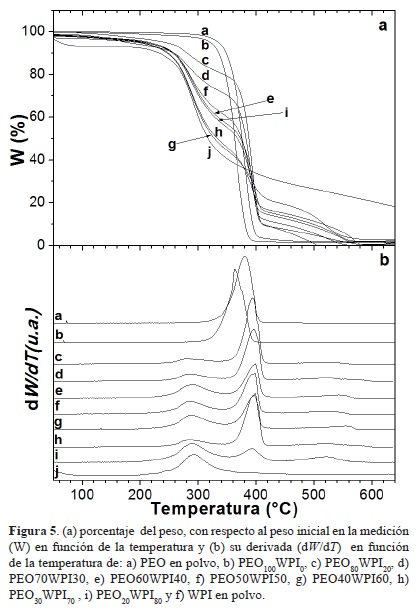
Las fibras con diferentes proporciones de PEO y WPI, exhibieron dos etapas principales de descomposición, que pueden ser más claramente distinguidas en las curvas de la primera derivada de TGA (Figura 5b). Las fibras compuestas mostraron las temperaturas de pérdida de peso características de sus componentes, es decir, de 250°C a 330 °C y 350°C a 400 °C. Es de notar que la temperatura de descomposición máxima de las fibras al combinarse con PEO/WPI en las diferentes proporciones no corresponde con exactitud a las temperaturas de sus componentes, sino que hay un desfasamiento (Figura 5b). Cuando el PEO es mezclado con WPI su pico de temperatura de degradación es más elevado que el de las fibras de PEO100WPI0 o el de PEO puro en polvo. Por su parte, el pico de temperatura máxima de degradación del WPI, cuando se combina con PEO, es menor al de su contraparte sin mezclar. Estas variaciones podrían atribuirse a la proporción en que estos compuestos se encuentran en las fibras y a sus diferentes capacidades de formar puentes de hidrógeno mejorando su estabilidad térmica. Las nanofibras obtenidas de las combinaciones PEO60WPI40 y PEO50WPI50 fueron térmicamente más estables.
4. Conclusiones
Se obtuvieron nanofibras de polímeros biodegradables y biocompatibles de PEOxWPI100-x para 0<x≤100, evaluando la influencia de la concentración de PEO en las propiedades morfológicas, estructurales y termogravimetricas de las nanofibras. Los diámetros de las nanofibras se mantuvieron muy cercanos al valor de 220 nm, para 40≤x≤100; incrementándose considerablemente para x <40, esto debido a la formación de "perlas". No fue posible la formación de nanofibras sin la adición de PEO en la solución a hilar. La cristalinidad de las nanofibras incrementó a medida que aumenta la cantidad de PEO en las nanofibras. Los análisis de TGA indicaron que las nanofibras obtenidas de las combinaciones PEO60WPI40 y PEO50WPI50 fueron térmicamente más estables.
Agradecimientos
Al Mtro. Francisco López Martínez por su apoyo técnico. Este trabajo fue financiado por el ICyT-DF bajo el proyecto PICSA11-18 y la SIP -IPN, bajo en proyecto 20131790. GRG agradece el apoyo financiero aportado del proyecto FOMIX-Yucatán No. 170120.
Referencias
[I]. K. Yong Lee, L. Jeong, Y. Ok Kang, S. Jin Lee, W. Ho Park, Adv. Drug Deliver Rev., 61, 1020 (2009). [ Links ]
[2]. T. J. Sill, H. A. von Recum, Biomaterials, 29, 1989 (2008). [ Links ]
[3]. A. Frenot, I. S. Chronakis, Curr Opin Colloid Interface Sci., 8, 64 (2003). [ Links ]
[4]. A. Fernandez, S. Torres-Guiner, J. M. Lagaron, Food Hydrocolloide, 23, 1427 (2009). [ Links ]
[5]. D. L. Woerdeman, P. Ye, S. Suresh, R. S. Parnas, G. E. Wnek, O. Trofimova, Biomacromolecules, 6, 707 (2005). [ Links ]
[6]. S. Alborzi, L. T. Lim, Y. Kakuda, Food Chemistry, 75, 100 (2009). [ Links ]
[7]. S. Moon, R. J. Farris, Polym. Eng. Sci., 49, 1616 (2009). [ Links ]
[8]. S. Torres-Giner, M. J. Ocio, J. M. Lagaron, Carbohydr. Polym., 77, 261 (2009). [ Links ]
[9]. J. Li, A. He, J. Zheng, C. C. Han, Biomacromolecules, 7, 2243 (2006). [ Links ]
[10]. S. Wongsasulak, K. M. Kit, D. J. McClements, T. Yoovidhya, J. Weiss, Polymer, 48, 448 (2007). [ Links ]
[11] . J. W. Lu, Y. L. Zhu, Z. X. Guo, P. Hu, J. Yu, Polymer, 47, 8026 (2006). [ Links ]
[12]. S. Moon, B. Y. Ryu, J. Choi, B. Jo, R. J. Farris, Polym. Eng. Sci., 49, 52 (2009). [ Links ]
[13]. A. Aluigi, C. Vineis, A. Varesano, G. Mazzuchetti, F. Ferrero, C. Tonin, Eur. Polym. J., 44, 2465 (2008). [ Links ]
[14]. A. C. Vega-Lugo, L. T. Lim, J. Polym. Sci. Pol. Phys., 50, 1188 (2012). [ Links ]
[15]. S. Dhawan, M. Varma, V. R. Sinha, Pharm. Technol., 29, 72 (2005). [ Links ]
[16]. S. Dhawan, K. Dhawan, V. R. Sinha, Pharm. Technol., 29, 82 (2005). [ Links ]
[17]. M. Demet, K. Fatmanur, I. Koichi, Y. Yusuf, O. Oguz, Polym Int. 53, 237 (2004). [ Links ]
[18]. C. C. Su, Y. H. Shen, Colloids & Surfaces A., 312, 1 (2008). [ Links ]
[19]. W. W. Sun, S. J. Yu, X. A. Zeng, X. Q. Yang, X. Jia, Food Res. Int., 44, 1052 (2011). [ Links ]
[20]. E. G. Mahamadou, S. Y. Xu, W. Zhang , J. Food Eng., 83, 521 (2007). [ Links ]
[21]. C. L. Mott, N. S. Hettiarachchy, M. Qi, J. Am. Oil Chem. Soc., 76, 1383 (1999). [ Links ]
[22]. D. Xu, X. Wang, J. Jiang, F. Yuan, Y. Gao, Food Hydrocolloids, 28, 258 (2012). [ Links ]
[23]. S. Kaya, A. Kaya, J. Food Eng., 43, 91 (2000). [ Links ]
[24]. M. B. Perez-Gago, J. M. Krochta, J. Food Sci., 66, 705 (2001). [ Links ]
[25]. Y. Yuan, Y. Gao, J. Zhao, L. Mao, Food Res. Int., 41, 61 (2008). [ Links ]
[26]. Y. Liu, G. Ma, D. Fang, J. Xu, H. Zhang, J. Nie, Carbohydr. Polym., 83, 1011 (2010). [ Links ]
[27]. M. Christopher, R. F. Burba, B. Grady, Electrochimica Acta, 53, 1548 (2007). [ Links ]

