











 text new page (beta)
text new page (beta) Spanish (pdf)
Spanish (pdf)
 Article in xml format
Article in xml format Article references
Article references
 Send this article by e-mail
Send this article by e-mail Cited by SciELO
Cited by SciELO  Similars in
SciELO
Similars in
SciELO 
 Permalink
Permalink1 Introducción
La síntesis de biocombustibles es una de las alternativas con mayor potencial para sustituir progresivamente el uso de combustibles fósiles no renovables, contribuyendo a mitigar el impacto ambiental que los mismos causan en el ecosistema, y a encontrar alternativas que minimicen los problemas de abasto y dependencia económica que implican los combustibles fósiles en el futuro cercano. Por esta razón existe un gran interés desde el punto de vista científico y tecnológico por diseñar nuevos procesos químicos que permitan la transformación de biomasa, un insumo renovable, en "combustibles verdes". Se hace notar que el éxito en aplicación comercial de los mismos está supeditado, entre otros factores, a que tengan propiedades físico químicas muy similares a las que exhiben los combustibles de origen fósil, de tal forma que se puedan emplear en una elevada proporción en los mismos motores de combustión.
Se pueden utilizar diversos tipos de biomasa para la síntesis de biocombustibles. A la fecha, el proceso más estudiado es la síntesis de biodiesel a partir de aceites que se extraen de oleaginosas, en particular de aceites no comestibles. Gradualmente, se ha incrementado el interés en el uso de aceites extraídos de microalgas y de biomasa residual de restaurantes y rastros municipales, para la síntesis de biodiesel. Numerosos trabajos han documentado la conversión de aceites vegetales comestibles y no comestibles en una mezcla de alcanos de la gama de compuestos que forman el diésel (C16-C18), la cual se denomina "diesel verde" (Kubičková, 2010). Notoriamente, este producto es totalmente compatible coneldiéseldeorigenfósilyelcombustibleque se comercializa es una mezcla de ambos productos, con proporciones creciente del diésel verde, cuyo uso reduce significativamente las emisiones de gases de efecto invernadero a medida que se incrementa la proporción de diésel verde en la mezcla (Sotelo- Boyás, 2011).
Por otra parte, varios estudios han reportado la conversión de biomasa en una mezcla de hidrocarburos que se identifica como "gasolina verde", y que tiene propiedades muy similares a gasolina de origen fósil (Twaiq y col., 2004; Ooi y col., 2005; Benson y col., 2009; Siswanto y col., 2008; Rao y col., 2010). A la fecha, los triglicéridos obtenidos a partir de oleaginosas son la principal materia prima para la producción de gasolina verde (Twaiq y col., 2004; Ooi y col., 2005; Benson y col., 2009; Siswanto y col., 2008; Rao y col., 2010; Kubičková,2010; Ong y col.,2010;Sotelo-Boyas, 2011). Particularmente, los aceites de palma y de canola se han transformado a gasolina verde a presión atmosférica y un rango de temperatura de 300-500 C, en presencia de catalizadores micro y mesoporosos con carácter ácido (Katikaneni y col., 1995; Ooi y col., 2005; Siswanto y col., 2008; Benson y col., 2009). Los productos de estas reacciones se encuentran en tres fases: gases, líquidos y sólidos (coque). Por lo general, los gases y el coque no presentan variaciones relevantes entre la mayoría de los trabajos reportados; sin embargo, en todos los casos la selectividad de los productos líquidos es función del tipo de catalizador utilizado. Así, se ha documentado que el uso de catalizadores ácidos microporosos (p.ej., HZSM-5 y HY) también promueve la formación de benceno, tolueno y o-, m-, p-xilenos (BTX). Por otra parte, los materiales ácidos mesoporosos (p.ej., MCM-41 y SBA-15) promueven preferentemente la formación de parafinas lineales y ramificadas (Corma y col., 1998; Twaiq y col., 2003; Benson y col., 2008; Ong y col., 2010). De esta forma, aun cuando los materiales microporosos exhiben mayor actividad bajo las mismas condiciones de reacción, los materiales mesoporosos presentan un mayor rendimiento hacia la fracción de gasolina (Twaiq y col., 2003; Zhao y col., 1996). Este hecho es particularmente importante desde la perspectiva industrial, ya que el contenido máximo permitido de BTX en la gasolina es menor al 1%/vol (pemex.ref.com), por lo que no es deseable la producción de una gasolina con las características de la mezcla obtenida cuando se usan catalizadores microporosos.
Se han propuesto diversos mecanismos para la conversión de aceites vegetales sobre aluminosilicatos cristalinos (zeolitas) y amorfos y se ha documentado las reacciones que ocurren únicamente por efecto térmico. Los elementos más relevantes de mecanismos más aceptados en la literatura se describen de forma muy general a continuación, ya que sobre esta base, en el actual trabajo se sustenta un mecanismo que complementa los descritos a continuación y que está avalado por las observaciones experimentales documentadas en este trabajo. Idem y col., (1997) estudiaron el rompimiento térmico del aceite de canola en un reactor continuo (P atm , 300-500 C, GHSV de 3.3-640 h−1) y encontraron un notorio efecto de la temperatura, la velocidad espacial y la presencia de vapor de agua. Estos autores propusieron que la conversión del aceite inicia por una etapa de pirólisis que genera una gran cantidad de etileno, el cual se consume en etapas subsiguientes que incluyen la generación de hidrógeno (H2) y el rompimiento secundario. Las reacciones de deshidrogenación son responsables de la producción de grandes cantidades de olefinas gaseosas C2-C4, las cuales son también una combinación de gases generados por reacciones de descarbonilación, descarboxilación y reacciones de rompimiento secundario. Finalmente, se obtienen hidrocarburos líquidos oxigenados, aromáticos y alifáticos. Por otra parte, Katikaneni y col., (1995) propusieron un mecanismo para el aceite de canola sobre sílica-alúmina amorfa. En este caso, el proceso inicia con una etapa térmica que rompe las moléculas de triglicérido en hidrocarburos alifáticos y moléculas oxigenadas pesadas. Estas moléculas participan en un proceso de rompimiento y desoxigenación para formar CO, CO2 y olefinas de cadena corta, que luego se oligomerizan para formar otras olefinas e hidrocarburos alifáticos. Estos hidrocarburos pueden generar una mezcla que también incluye hidrocarburos aromáticos mediante un mecanismo complejo de reacciones que incluyen isomerización, alquilación y aromatización o hidrocarburos gaseosos resultantes de rompimiento catalítico. El modelo también incluye dos posibles rutas de formación de coque: la polimerización de aromáticos y la condensación de las moléculas de aceite.
Uno de los primeros mecanismo de rompimiento catalítico de aceite en zeolitas fue propuesto por Prasad y col., (1986) e incluye 6 etapas principales: i) desoxigenación, que consiste en la descarboxilación y la deshidratación de la molécula de triglicérido en la superficie externa del catalizador; ii) el rompimiento primario de las moléculas de mayor tamañoatravésdelmecanismodeabstracciónde hidruro; iii) la difusión de las cadenas más cortas a través de los poros del catalizador; iv) las reacciones de rompimiento secundario para formar olefinas; v) la aromatización, isomerización y transferencia de hidrógeno para formar parafina y olefinas; vi) la formación de coque. Este mecanismo se ha complementado progresivamente con diversas contribuciones. Katikaneni y col., (1995) propusieron que en la etapa (iv) las parafinas más cortas y las olefinas se oligomerizan, ciclizan e isomerizan para formar parafinas y olefinas C2 -C20, así como compuestos aromáticos. Además, añadieron una última etapa (vii), un proceso en competencia para la formación directa de coque a partir de la molécula del triglicérido. Posteriormente, Idem y col., (1997) postularon que la etapa (i) es independiente de la naturaleza del catalizador, y que en la etapa (iv) la selectividad de forma de las zeolitas obstaculiza el rompimiento secundario de las cadenas cortas en los poros del catalizador. Por su parte, Leng y col., (1999) identificaron las etapas que favorecen a la fracción gasolina; sugirieron que las olefinas formadas en la etapa (iv) se oligomerizan para producir una mezcla de alcanos y alquenos pesados, formando gasolina, queroseno y diesel. Sobre las premisas anteriores, Benson y col., (2009) presentaron un mecanismo más completo para el rompimiento catalítico de aceites vegetales sobre zeolitas, que incluye las principales etapas y familias de reacciones antes descritas. De hecho, la contribución más importante de Benson y col. (2009) fue sugerir dos posibles rutas para iniciar el proceso. Una ruta (1) propone la adsorción de las moléculas de un aceite insaturado en el sitio ácido de la zeolita; después, el mecanismo ocurre a través de las etapas ya descritas. La otra ruta (2) propone en primer término la liberación de los ácidos grasos de la molécula del triglicérido y su posterior procesamiento, que corresponde a la conversión de las moléculas oxigenadas pesadas referidas previamente en la etapa (ii).
No obstante los alentadores avances logrados a la fecha, algunos de los retos importantes por resolver en la conversión de biomasa en gasolina verde incluyen la optimización de la formulación ácida de los catalizadores y de las condiciones experimentales utilizadas, que permitan maximizar la actividad y selectividad hacia la fracción de gasolina. Además, se debe continuar explorando alternativas a los triglicéridos típicamente utilizados como materia prima. A este respecto, una variante que recibe ahora mucha atención es el uso de aceites vegetales no comestibles, extraídos de cultivos extensivos, como es el caso de diferentes variedades de Jatropha e higuerilla. De hecho, estos cultivos ya se han utilizado comercialmente para la obtención de aceite que luego se convierte en biodiesel (Nieves Soto y col., 2012; Soto León y col, 2014); sin embargo, no se ha documentado su potencial en la síntesis de otros combustibles verdes. Otra materia prima alternativa de particular interéses la biomasa residual generada en diversos procesos de fermentación industrial. En la actualidad, existen una diversidad de procesos importantes desde la perspectiva industrial y económica que implican fermentaciones y, que una vez extraído el producto, generan biomasa residual que no recibe un tratamiento o aprovechamiento adecuado. Esta situación se identifica típicamente en la industria de alimentos (p. ej.,lácteos,nutracéuticos,licores,vinos,cervezas), la farmacéutica (p. ej., antibióticos, especialidades, proteínas) y la de energéticos (p. ej., biometano, bioetanol, butanol); más recientemente, es también el caso de la industria que busca reemplazos "verdes" para detergentes, limpiadores y empaques (Wenda y col., 2011; Iqbal y col., 2013). Notoriamente, en todos estos procesos está implícita la generación de biomasa (Nielsen y col., 2003), la cual representa un nicho de oportunidad para desarrollar nuevos procesos que permitan su transformación en productos de alto valor agregado, como puede ser el caso de combustibles verdes. A la fecha, existen limitados estudios sobre el uso de este tipo de biomasa, o de moléculas modelo similares, que faciliten el entendimiento y la optimización de su empleo en la síntesis de combustibles verdes (Mrad y col, 2013; Mussatto, 2013; Xiao y Anderson, 2013).
Con base a lo anterior, el objetivo de este trabajo fue evaluar la conversión de lecitina en combustibles verdes, como un sistema modelo para determinar el posible uso de fosfolípidos presentes en la biomasa residual de diversos tipos de industrias, como materia prima alternativa en la síntesis de combustibles verdes. El sistema de reacción implementado para la conversión de lecitina se validó previamente mediante la conversión de aceite de canola sobre H-MCM41, a 400 C y presión atmosférica. Los estudios con este sistema permitieron identificar condiciones experimentales apropiadas para la conversión de lecitina, así como establecer un mecanismo a partir del cual se tiene un mejor entendimiento de los procesos elementales que ocurren en el sistema de reacción. Esta información puede facilitar la identificación de las condiciones óptimas para promover más selectivamente la síntesis de productos de interés, así como determinar el tipo y la configuración de sitios ácidos requeridos para este propósito. Sobre estas bases se puede favorecer entonces el desarrollo de procesos adecuados para para la producción de combustibles verdes.
2 Metodología y materiales
El sistema de reacción utilizado fue un reactor tubular construido en acero inoxidable 316, de 40 cm de longitud y 0.42 cm de diámetro. La temperatura del reactor se controló utilizando un sistema integrado por un horno de resistencia eléctrica con elemento cerámico, un controlador Nimbus N1100 y un termopar tipo K. La alimentación al reactor consistió de una mezcla gas/líquido que se evapora a la entrada del reactor y forma una mezcla gaseosa uniforme antes de entrar en contacto con el catalizador, el cual se cargó en la parte central del reactor. En este estudio preliminar se utilizó He en la alimentación, cuyo flujo se reguló entre 20 y 100 cc/min usando controladores de flujo másico MKS. El aceite de canola utilizado fue marca Maravilla, y la lecitina de soya grado alimenticio fue de la marca Pronat Ultra. Los líquidos se alimentaron al reactor a un flujo de 0.005 hasta 0.055 cc/min, usando una bomba de infusión KD Scientific. La descarga del reactor se conectó a una trampa de condensados a temperatura ambiente y presión atmosférica; los gases y vapores no condensados en la descarga de la trampa se dirigieron a un sistema de cromatografía de gases para su análisis en línea.
El monitoreo en línea de los gases producidos en la reacción se realizó usando dos cromatógrafos de gases (CG) Agilent 6850 conectados en serie; un cromatógrafo, equipado con un detector de conductividad térmica (TCD) y una columna HP- PlotQ, se usó para la detección de H2, CO, CO2 y H2O. En este caso se utilizó He como gas acarreador (Praxair, grado 4.6Z), y la columna se mantuvo a 40 C por 6 min, después se calentó a 20 C/min hasta 120 C, donde se mantuvo durante 1 min. Un segundo cromatógrafo, equipado con un detector de ionización de flama (FID) y una columna Al/S, se usó para la detección de parafinas C1-C6, isoparafinas C3-C5 y olefinas C2-C5. En este caso se utilizó N2 como gas acarreador (Praxair, grado 5.0), y la columna se mantuvo a 35 C por 0.5 min, luego se calentó a 15 C/min hasta 100 C, y después a 10 C/min hasta 180 C. Los factores de calibración de los gases antes indicados se obtuvieron de forma experimental, utilizando mezclas estándares adquiridas a Praxair.
Por otra parte, el líquido acumulado en la trampa de condensados se colectó en lotes correspondientes a una hora de operación y se analizó por tres métodos.
i) Derivatización de los ácidos grasos a metil-ésteres, utilizando tri-ricinoleína de Sigma Aldrich como estándar interno, para determinar la conversión total en las muestras con lípidos. El análisis se realizó en un CG marca Agilent, modelo 6850 FID, equipado con una columna HP-1 y utilizando N2 como gas acarreador. En estos análisis, la temperatura del puerto de inyección se mantuvo a 300 C y la del detector a 250 C; la columna se mantuvo a una temperatura inicial de 60 C y luego se calentó con una rampa de 15 C/min hasta 260 C, donde se mantuvo durante 3 min.
ii) Derivatización con N-metil-N-(trimetilsilil) trifluoroacetamida (MSTFA) adquirida de Sigma- Aldrich, para determinar la conversión de las muestras que contenían ácido oleico. El análisis se realizó usando el equipo y las condiciones reportadas en el caso anterior.
iii) La identidad de los hidrocarburos en la mezcla de condensados a la descarga del reactor se determinó en un CG equipado con un espectrómetro de masas Agilent 6890 y una columna DB5-ms. Posteriormente, la cuantificación de estos productos se realizó rutinariamente usando un CG equipado con un FID y con una columna HP-1, con N2 como gas acarreador. En estos análisis, la temperatura del puerto de inyección se mantuvo a 300 C y la del detector a 250 C; la temperatura de la columna se mantuvo inicialmente a 40 C por 5 min, después se calentó a 10 C/min hasta 170 C, manteniéndose a esta temperatura durante 10 min; después, se calentó secuencialmente con rampas de 5 C/min hasta 200 C, a la cual se mantuvo durante 5 min, y de 3 C/min hasta 325 C, temperatura a la cual se mantuvo durante 5 min.
Los factores de conversión de los metilésteres se determinaron experimentalmente, mientras que la proporción entre las diferentes fracciones de productos líquidos se realizó a partir de la relación de las áreas calculadas por el integrador de la señal del CG. La Tabla 1 incluye los rangos de temperatura en los cuales se distribuyeron los productos líquidos analizados. La naturaleza de cada fracción se asignó de acuerdo a la distribución de áreas en el cromatograma. A manera de control, el análisis de algunas muestras se corroboró con la cuantificación realizada con los datos del detector de espectrometría de masas.

Para las pruebas de reacción se usó como catalizador el tamiz molecular MCM41 de la compañía Tianjin, con una relación de Si/Al 25:1. El catalizador se sometió a un intercambio iónico con NH4Cl, a 80 °C, en tres etapas sucesivas (Theng y col., 1968). Después del intercambio iónico, y su posterior calcinación, el catalizador identificado como H-MCM41, se caracterizó por difracción de rayos X de ángulo amplio (WAX) y pequeño (SAX). Para el análisis WAX se usó un equipo Rigaku Dmax 2200 con radiación de tubo de cobre a 36 kV y 30 mA; por otra parte, el análisis de SAX se llevó al cabo en un equipo Nanostar marca Bruker, utilizando radiación de cobre a 36 kV. Morfológicamente, el catalizador se caracterizó mediante fisisorción de N2 a 77 K, usando un equipo ASAP 2020 de Micromeritics, para evaluar el área superficial y el volumen de poros. Por otra parte, la validación de los sitios ácidos se realizó con la técnica acoplada de adsorción de piridina y espectrometría infrarroja con transformada de Fourier, en un equipo Thermo Scientific, modelo Nicolet 6700, con una resolución de 4 cm−1 y 100 barridos. Previo alanálisis, la muestra de H- MCM-41 se sometió a un tratamiento con O2 a 350 C. Durante el análisis la muestra se expuso, secuencialmente, a una corriente de He saturado con piridina a diferentes temperaturas entre 200 y 350 C, y después se obtuvo el espectrograma de infrarrojo correspondiente a temperatura ambiente. Con base a estudios previos reportados en la literatura (Twaiq y col., 2003; Ooi y col., 2005), en este trabajo se propuso realizar las pruebas de reacción del aceite de canola y de la lecitina sobre H-MCM41 se realizaron a 400 C y presión atmosférica, a diferentes valores de la velocidad específica espacial, weight hourly space velocity (WHSV), la cual se calculó del cociente entre la masa del catalizador cargado al reactor y el flujo másico de la alimentación a la entrada del reactor. Para el aceite de canola se utilizaron valores de WHSV de 0.1, 1.0, 2.5 y 10 h−1, y para la lecitina los valores de WHSV fueron 1.0 y 2.5 h−1, que están en el rango de otros trabajos previamente reportados (Twaiq y col., 2003; Ooi y col., 2005). La conversión y el rendimiento carbón-mol de los productos se definieron de acuerdo a las siguientes ecuaciones:
 (1)
(1)
 (2)
(2)
3 Resultados
3.1 Caracterización del catalizador
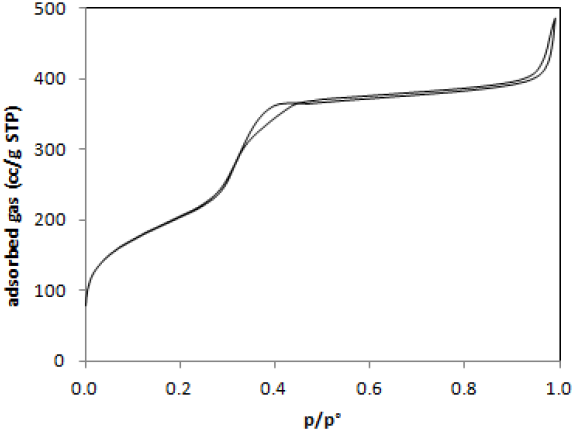
Los resultados de SAX y WAX se presentan integradamente en la Figura 1. Estos resultados revelaron que el material presenta planos cristalinos en ángulos de 2Θ de 2, 4 y ~22, congruentes con los planos 100, 110 y 200, respectivamente. La presencia de estos planos se asoció a una estructura en forma de panal, congruente con lo reportado en la literatura (Ryoo y col., 1997; Sonwane y Bathia, 1999; Antonakou y col., 2006). Por otra parte, la Figura 2 muestra la isoterma de adsorción de N2 sobre H-MCM41 a 77 K. La isoterma presentó histéresis, comportamiento típico de materiales mesoporosos altamente ordenados del tipo MCM41, con un incremento en la adsorción de N2 a presiones relativas entre 0.3 y 0.4(Iliopoulou y col.,2007). Elárea superficial de la H-MCM41 se calculó usando el modelo BET en 720.8 m2/g, la cual está en el intervalo característico de este tipo de materiales (Zhao y col, 1996; Tompset y col., 2005). Por otra parte, el diámetro de poro se determinó usando la isoterma de desorción con el modelo BJH, encontrando un tamaño promedio de poro de 2.5nm ±0.4nm, que corresponde a materiales mesoporosos.

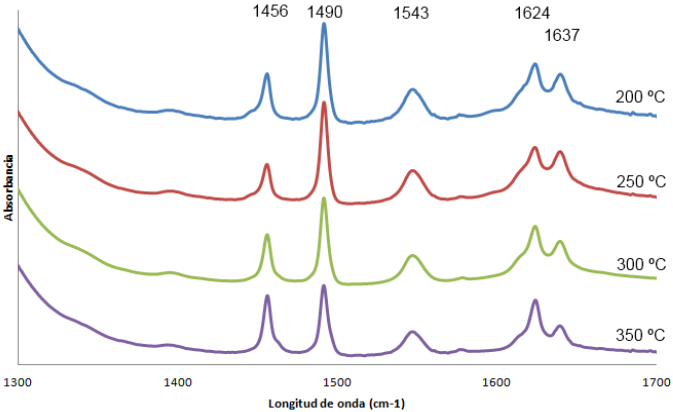
Por otra parte, la Figura 3 muestra el espectro de infrarrojo obtenido después de la adsorción de piridina en H-MCM41, a temperaturas entre 200 y 350 C. Tomando como referencia las frecuencias de infrarrojo características de la molécula de piridina adsorbida en diferentes tipos de sitios ácidos (Chackraborty, 1999), se identificó la presencia de sitios tipo Brönsted (1543 y 1637 cm-1) y de sitios tipo Lewis (1456 cm-1); también se identificó la banda común que corrobora la presencia de ambos tipos de sitios en la muestra (1490 cm-1); los resultados de la Figura 3 sugieren que los sitios identificados son estables. La caracterización del tipo de acidez de la muestra de H-MCM41 fue similar a la reportada por Twaiq y col. (2003), quienes identificaron la presencia de ambos tipos de sitios ácidos, Brönsted y Lewis, en un aluminosilicato mesoporoso que mostró actividad catalítica para el rompimiento de aceite de palma. Típicamente, los sitios Brönsted promueven la familia de reacciones asociadas al rompimiento catalítico (es decir, iniciación por adsorción de olefinas, oligomerización/rompmiento β, isomerización, transferencia de hidruro), y los sitios Lewis pueden favorecer preferentemente la iniciación por protonación de alcanos (Sanchez-Castillo y col., 2002)
3.2 Perfil de ácidos grasos de los reactivos
El análisis FAME del aceite de canola utilizado en este estudio indicó que no contiene aditivos ni nutracéuticos (Tabla 2). Losresultadosdelanálisis fueron congruentes con lo reportado en la literatura para este aceite (canolacouncil.org). De igual manera, el análisis FAME de la lecitina indicó que la composición de los ácidos grasos fue similar a la reportado para la lecitina de soya (Huang y col., 2007).
Tabla 2 Composición determinada para el aceite de canola y la lecitina por el método FAME.
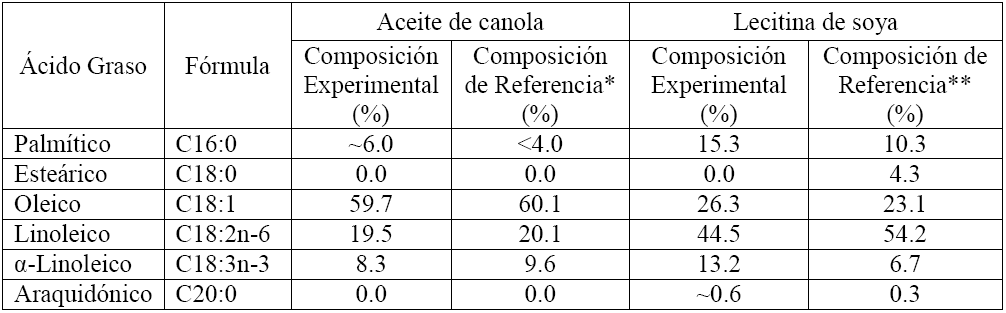
*The canola council of Canada (canolacouncil.org); **Huang y col., 2007.
3.3 Conversión catalítica
En primer término se reporta la conversión catalítica del aceite de canola, con el objetivo de facilitar la validación del sistema de reacción construido mediante su comparación con otros resultados reportados en el literatura. Posteriormente, se presentan los resultados de la conversión catalítica de la lecitina, para los cuales no se encontraron reportados estudios similares.
3.3.1. Aceite de canol
3.3.1.1 Estabilidad
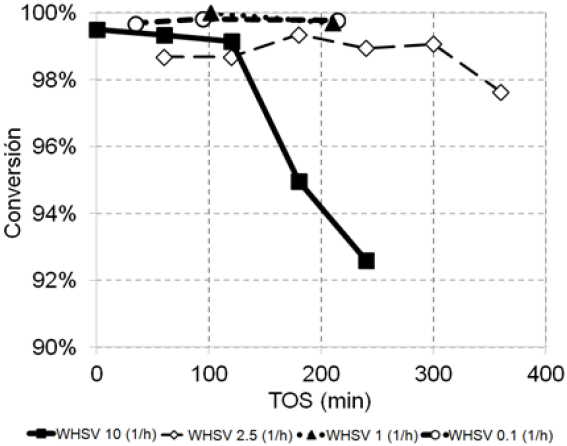
La Figura 4 muestra la conversión total del aceite de canola sobre H-MCM-41 a presión atmosférica, 400 C, y diferentes valores de WHSV entre 0.1 y 10 h−1. En los experimentos a WHSV de 0.1 y 1 h−1, la conversión se mantuvo prácticamente invariante durante 4 h. En el experimento a una WHSV de 2.5 h−1 la actividad empezó a disminuir a partir de 300 min, y en el experimento a una WHSV de 10 h−1 la conversión disminuyó ligeramente en los primeros 100 min de reacción, y luego decayó hasta 92% después de 240 min de operación. Al terminar cada uno de los experimentos, se observó una materia carbonosa depositada sobre la superficie del catalizador recuperado, lo cual indicó que la desactivación en el catalizador ocurrió al formarse coque en la superficie. La cantidad de coque se relacionó con la cantidad de reactivo tratada. De acuerdo a los datos de todas las reacciones, HMCM-41 presentó desactivación después de haber transformado 5 g de aceite de canola, que equivale a 16.7 veces la masa de catalizador cargada para la reacción.
3.3.1.2 Rendimiento en fase gas: Estabilidad y efecto WHSV
En la Figura 5 se presentan los datos de rendimiento 4 hacia CO y CO2 en función del tiempo de reacción a diferentes valores de WHSV: 10, 2.5, 1.0 y 0.1 h−1. El rendimiento hacia CO y CO2 se monitoreó en línea, analizando la mezcla de gases arrastrados desde la trampa de condensados a la salida del reactor cada 30 min. En todos los casos, el rendimiento hacia CO fue mayor que el rendimiento hacia CO2. Además, se encontró que la WHSV tuvo un efecto en la relación CO/CO2.
a) En los experimentos realizados a WHSV de 10 y 2.5 h−1, el rendimiento para CO se mantuvo entre 1.3 a 1.5%, y el de CO2 entre 0.6 y 0.75 %. En estos experimentos la razón de rendimientos de CO/CO2 fue aproximadamente igual a 2.
b) Por otra parte, para los experimentos realizados a WHSV de 1.0 y 0.1 h−1, el rendimiento para CO se mantuvo entre 1.0 y 1.2 %, y la relación de rendimiento de CO/CO2 en ambos experimentos fue aproximadamente igual a 1
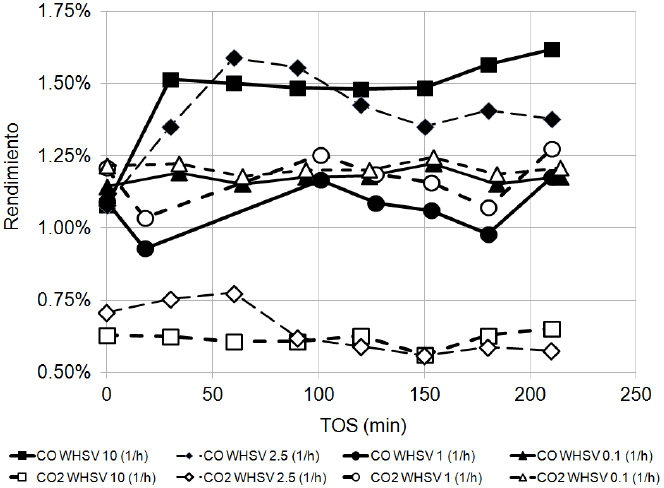
Fig. 5 Efecto de WHSV en el rendimiento carbón-mol de CO y CO2 en el rompimiento catalítico del aceite de canola sobre H-MCM-41 a 400°C y Patm.
Por otra parte, las Figuras 6 y 7 muestran, respectivamente, el rendimiento carbón-mol hacia parafinas y olefinas gaseosas identificadas durante el rompimiento catalítico del aceite de canola a 400 °C, 1 atm y WHSV 2.5 h−1. Diversos autores han reportado estos gases como característicos de procesos de rompimiento secundario (Kissin, 2001 y Abbott y col., 1988). Los resultados muestran que el rendimiento carbón-mol de estos productos en fase gas fue menor a 0.3% en todos los casos. Para las parafinas, el rendimiento carbón-mol fue de 0.15%, 0.16% y 0.20% para n-butano, propano y etano, respectivamente. Para las olefinas, los mayores rendimientos carbón-mol fueron: cis-2-buteno (0.25%), trans-2-buteno (0.15%)

Fig. 6 Rendimiento carbón-mol hacia parafinas gaseosas para el rompimiento catalítico del aceite de canola sobre H-MCM41 a 400 °C, P atm y WHSV 2.5h−1.
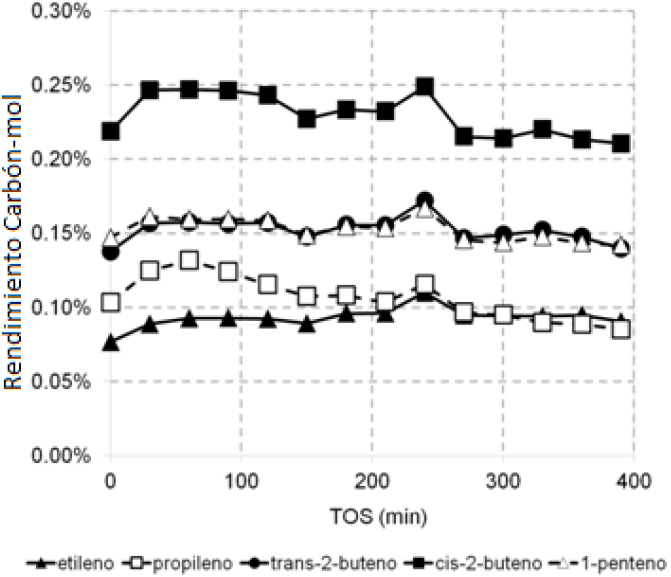
3.3.1.3 Rendimiento en fase liquida: Estabilidad y efecto WHSV
La Figura 8 muestra la composición de los productos líquidos del rompimiento catalítico del aceite de canola sobre H-MCM41 a diferentes valores de WHSV, de acuerdo a las fracciones indicadas en la Tabla 1. El análisis de los productos líquidos presenta 4 diferentes grupos de productos: hidrocarburos del tipo gasolina, hidrocarburos del tipo diesel, hidrocarburos con más de 18 átomos de carbono y ácidos grasos libres (principalmente C18). Los resultados indican la presencia de ácidos grasos libres en los experimentos a WHSV de 10 y 2.5 h−1, los cuales no se detectaron en los experimentos a WHSV de 1.0 y 0.1 h−1. En estos experimentos, las fracciones de gasolina y diésel no presentaron una dependencia específica respecto a la WHSV.
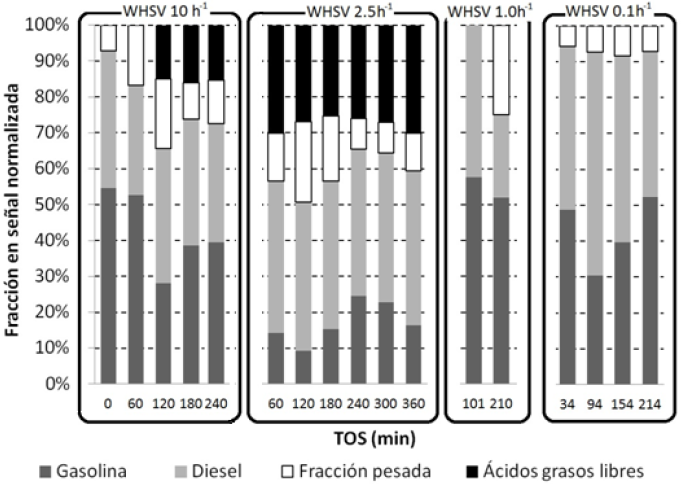
Fig. 8 Distribución de los productos líquidos del rompimiento catalítico de aceite de canola sobre H-MCM41 a 400°C, P atm y diferentes valores de WHSV.
Para WHSV de 10 h−1, la masa de hidrocarburos líquidos producidos disminuyó con el tiempo de reacción ya que comenzaron a producirse ácidos grasos (preferentemente oleico) en lugar de parafinas C1 -C18 u olefinas C2 -C6. En la mezcla de productos líquidos, se identificó ácido oleico,y este representó hasta aproximadamente el 20% de la masa líquida recuperada. Por otra parte, para WHSV de 2.5 h−1 la mezcla de hidrocarburos líquidos más abundante fue la fracción diésel (aproximadamente 40%),seguida por el ácido oleico (aproximadamente 30%) a lo largo de todo el tiempo de reacción estudiado. En estas condiciones de operación (WHSV=2.5 h−1) se observó que la selectividad para los productos líquidos es relativamente invariante, en forma análoga a lo observado para los productos gaseosos (Figuras 7 y 8). Adicionalmente, para los experimentos a WHSV de 10 y 2.5 h−1, se detectó el ácido 9-octadecenoico, tanto en la conformación E como en la conformación Z, particularmente notorio porque el aceite alimentado al reactor solo contenía la conformación Z.
En el experimento a WHSV de 1.0 h−1, la fracción líquida predominante fue gasolina (aproximadamente 50%); mientras que para el experimento a WHSV de 0.1 h−1 las fracciones predominantes fueron diesel (entre el 40 y 60%) y gasolina (entre el 40 y 50%). En estas dos últimas condiciones de operación no se detectó la producción de ácido oleico libre.
Al analizar el efecto de las diferentes WHSV, no se encontró una tendencia definida para la selectividad hacia gasolina y diesel en el intervalo de condiciones estudiado. Sin embargo, la aparición de ácidos grasos a WHSV de 10 y 2.5 h−1 se relacionó con el rompimiento de la molécula del triglicérido. Este rompimiento puede suceder en etapas, dentro de las cuales la liberación de los ácidos grasos ocurre rápidamente, y la formación de la fracción gasolina ocurre en etapas posteriores. Asimismo, a una WHSV de 0.1 h−1 se favoreció notoriamente la producción de coque, disminuyendo la actividad y estabilidad del catalizador.
3.3.2 Lecitina
3.3.2.1 Actividad
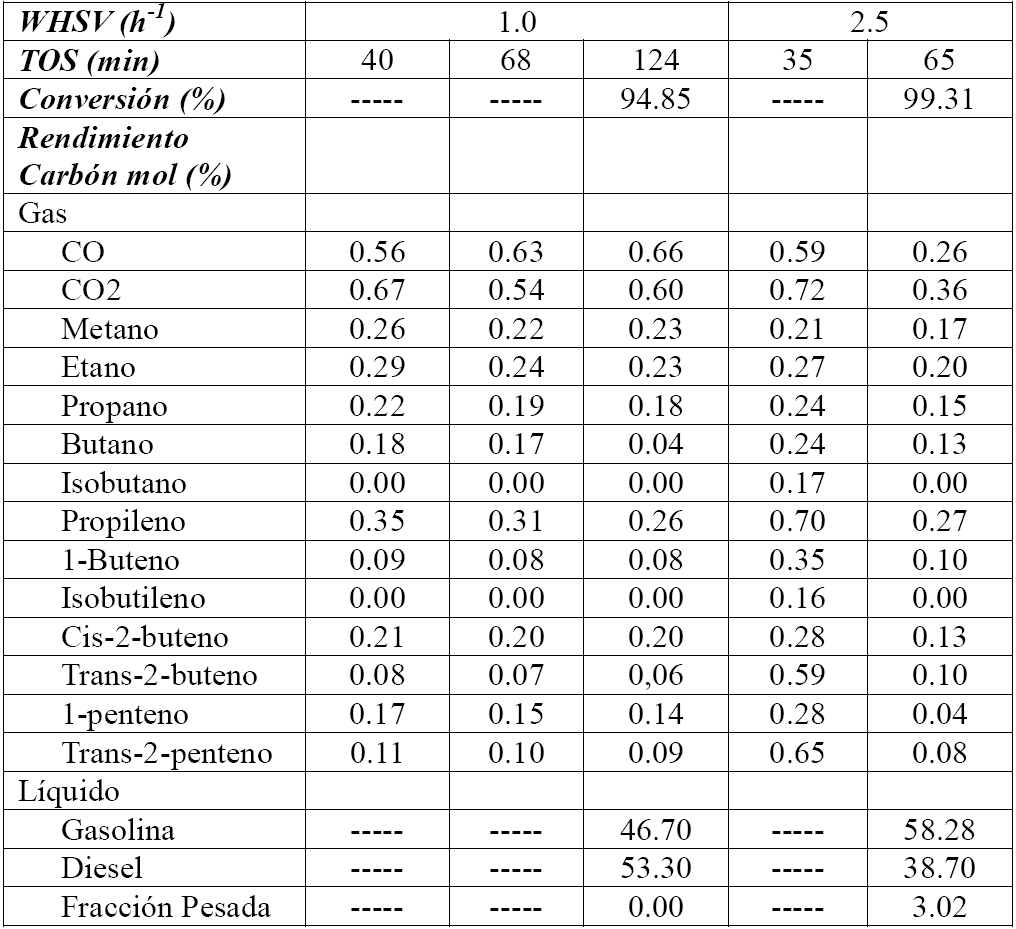
La actividad de la H-MCM41 para el rompimineto de la lecitina se evaluó a las condiciones identificadas como preferentes a partir de los experimentos de aceite de canola. Notoriamente, el catalizador H-MCM41 mostró actividad para el rompimiento catalítico de la lecitina a 400°C, presión atmosférica y WHSV de 1.0 y 2.5 h−1. La Tabla 3 muestra que para estos experimentos la conversión total fue de 94.9 y 99.3%, respectivamente, que fue similar a la conversión correspondiente al aceite de canola (>90%). En este caso, no se observó un efecto de la WHSV en la actividad o la selectividad en el intervalo de condiciones estudiado. Se hace notar que durante la conversión de lecitina el reactor se presurizó, sugiriendo el bloqueo del sistema de reacción posiblemente debido a la formación de un sólido. Para el experimento a WHSV de 2.5 h−1, el sistema se bloqueó aproximadamente a los 60 min después de iniciar la reacción (~2 g de masa transformada) y para el experimento a 1.0 h−1, el sistema se bloqueó a los 120 min después de iniciar la reacción (~2 g de masa transformada). El tiempo de reacción se denota en este trabajo como "time on stream" (TOS). Esta información indicó que el bloqueo se presentó en cada experimento después de tratar la misma cantidad de materia prima. Una vez desensamblado el reactor, se encontró un sólido depositado sobre la superficie del catalizador, que posiblemente bloqueó la inyección de reactivo al sistema. Este sólido presentó características de apariencia (color) y mojabilidad (hidrofóbico) diferentes al sólido depositado durante el rompimiento del aceite de canola. Se ha reportado en la literatura (Nawar,1969; Nawar,1984) que los fosfolípidos son termosensibles, y que el enlace que une la estructura de glicerol con el ácido fosfórico es energéticamente débil comparado con el resto de los enlaces en la molécula. Cuando este enlace se rompe, genera dos estructuras: un aminoácido y dioleína. Este hecho sugiere que, bajo las condiciones de reacción, la lecitina se degradó térmicamente al entrar en el reactor generando fosfatidilcolina y dioleína. Dado que la fosfatidilcolina es sólida a condiciones de presión y temperatura ambientales, es posible que el sólido formado haya sido fosfatidilcolina, y subproductos de su propia degradación térmica. Esta propuesta se soportó por el hecho de que no se detectaran aminas gaseosas ni fósforo en los productos líquidos cuando se inyectaron en el CG/EM.
3.3.2.2. Selectividad
La Tabla 3 muestra que los productos gaseosos de mayor abundancia fueron CO y CO2, con un rendimiento cercano a 0.6% para ambos. La variación de WHSV de 1.0 a 2.5 h−1 no tuvo un impacto significativo entre los rendimientos de CO y CO2. Al evaluar el rendimiento carbón-mol hacia parafinas e isoparafinas C1-C6 , así como de olefinas C2 -C5 se observa que las olefinas (etileno, 1-butileno, trans-2-butileno y trans-2-penteno) fueron ligeramente más abundantes que las parafinas (etano, propano y n-butano) hasta en 0.5%. Sin embargo, esta diferencia no pareció ser significativa y el rendimiento total hacia gases fue menor a 3.5% y a 5.5% para los experimentos a WHSV de 1.0 y 2.5 h−1, respectivamente. En general, en las condiciones de estudio, no se encontró efecto de la WSHV en la formación de todos los productos gaseosos. Se hace notar que las parafinas etano, propano y n- butano fueron las de mayor rendimiento, similar a lo observado en el rompimiento catalítico del aceite de canola. Asimismo, las olefinas trans-2-penteno y cis-2-buteno fueron también comunes para ambos sistemas. Estas similitudes sugieren que los procesos de transformación de la lecitina y del aceite de canola sobre H-MCM41, a las condiciones estudiadas, son químicamente similares, ya que los gases generados en la reacción son representativos de los procesos que ocurren en la superficie del catalizador.
Por otra parte, el análisis de la fase líquida indicó que los productos de reacción fueron principalmente las fracciones de gasolina y diésel (enconjunto> 97%), siendo marginal la fracción pesada (< 3%). La fracción de gasolina (46.7%) y diésel (53.3%) fueron similares a una WHSV de 1.0 h−1, y la fracción de gasolina fue 12% mayor en el experimento a WHSV de 2.5 h−1; además, no se detectó la presencia de ácidos grasos en la mezcla de productos líquidos. De esta forma, el efecto de la WHSV sobre la conversión y el rendimiento hacia la fracción gasolina fue diferente al observado en el aceite de canola. Esto se puede atribuir a la deposición del sólido generado durante el rompimiento de la lecitina, que parece inhibir las reacciones de rompimiento secundario, promoviendo en primer término la desactivación del catalizador. De ser el caso, una WHSV menor inhibirá en mayor medida los procesos catalíticos que ocurren en la superficie de H-MCM41, limitando los procesos de oligomerización/escisión β, isomerización y transferencia de hidruros necesarios para la formación de la fracción de gasolina. La ausencia de ácidos grasos en la mezcla de productos líquidos se asoció a que una molécula de triglicérido posee tres ácidos, mientras que una molécula de lecitina sólo posee dos; esto hace que se procese una menor cantidad de ácidos grasos y, en consecuencia, ocurra el rompimiento efectivo de todos ellos.
4 Discusión
4.1 Resultados experimentales
De acuerdo con la literatura, la producción de la fracción gasolina a partir de aceites vegetales es una función de la morfología del catalizador (Idem y col., 1997; Twaiq y col., 2003; Twaiq y col., 2004; Ooi y col., 2005), del tipo y número de sitios (Katikaneni y col., 1996; Idem y col., 1997; Chen y col., 2010; Ngo y col., 2010), de la relación C/O en la alimentación (Siswanto y col., 2008) , y de la temperatura (Katikaneni y col., 1996; Siswanto y col., 2008) y la WHSV (Prasad y col., 1986; Siswanto y col., 2008).
En relación a las características morfológicas del catalizador, el área superficial solo tiene efectos marginales, menores a 5%, en el rompimiento catalítico de aceites vegetales (Idem y col., 1997; Twaiq y col., 2003). Sin embargo, se ha documentado que el tamaño de poro si tiene un efecto relevante en la conversión y la selectividad de los productos (Twaiq y col., 2004), y se debe hacer notar que este efecto está acoplado también a la estructura del aluminosilicato utilizado. Trabajos previos del rompimiento catalítico del aceite de canola han mostrado que las zeolitas (HZSM-5, H-MOR, H-Y) presentan mayor actividad, mayor selectividad hacia gases y selectividad de forma para los hidrocarburos líquidos (principalmente hacia tolueno y xilenos) que los aluminosilicatos amorfos (silicalita, sílica-alúmina, aluminio pilareado), los cuales, en contraparte, favorecen la formación de parafinas líquidas (Katikaneni y col., 1995). En las condiciones usadas en este trabajo, la conversión de aceite de canola sobre H-MCM41 dio lugar preferentemente a la formación de hidrocarburos líquidos que en función de su longitud de cadena se identificaron en el rango la gasolina verde o el diésel verde. Como se discute a detalles en una sección posterior, la proporción de cada uno de estos productos fue dependiente de la WHSV; sin embargo, en la condiciones estudiadas, la distribución de productos observada fue consistente con lo reportado por otros autores para materiales similares (Twaiq y col., 2004; Ooi y col., 2005).
Por otra parte, se ha documentado diversos tipos de sitios activos en la producción de combustibles verdes a partir de aceites vegetales, incluyendo catalizadores con sitios ácidos (HZSM-5, sílica-alúmina), con sitios básicos (óxido de magnesio, óxido de calcio) e incluso en otros materiales (silicalita y sílica) (Idem y col., 1997). Para un mismo catalizador ácido, el tipo y el número de sitios determinan la actividad y selectividad. Como se indicó, los sitios ácidos Brönsted, evidenciados en la Figura 3 por las bandas a 1543 y 1637 cm−1, son activos preferentemente para la familia de reacciones asociadas al rompimiento catalítico del aceite vegetal y a la producción de la fracción gasolina (Twaiq y col.,2003; Sanchez-Castillo y col, 2002). Como se detalla posteriormente, en las condiciones de este trabajo, se postula que los sitios Brönsted del catalizador H-MCM-41 son también los responsables de la etapa de iniciación del rompimiento catalítico. Por otra parte no se evidenció en las condiciones de reacción utilizadas el efecto catalítico de los sitios Lewis, para lo cual se deberán hacer estudios adicionales en otras condiciones de reacción (p. ej., a mayor temperatura).
Diversos trabajos en la literatura han reportado el efecto de la composición de la materia prima, incluyendo el estudio de mezclas con ácidos grasos libres, o la comparación con solo ácidos grasos libres (Ooi y col., 2005; Benson y col., 2009). Sin embargo, no se encontró documentado el estudio de moléculas modelo asociadas a fosfolípidos. Debido a la diversidad de las composición de las mezclas usadas como materia prima las comparaciones no son sencillas pero, en general, es claro que la composición de ácidos grasos, sin importar si se alimentan como ácidos grasos libres o como ésteres unidos a una molécula de glicerol (triglicérido), tiene un notorio impacto en las reacciones superficiales catalíticas. Un análisis detallado de los resultados en la literatura indica que los enlaces insaturados en las cadenas de carbono de los ácidos grasos pueden tener un notorio efecto en el proceso. Por ejemplo, Ooi y col. (2005) mostraron que una alimentación de ácidos grasos insaturados puede extender el tiempo que el catalizador permanece activo, si se compara con una materia prima rica en ácidos grasos saturados. En el presente trabajo, el aceite de canola y la lecitina presentaron un 87.5% y 84% de ácidos grasos insaturados, respectivamente. De esta forma, se propuso que el proceso de rompimiento catalítico para el aceite de canola y la lecitina eran similares, dado que la composición de los ácidos grasos insaturados fueron similares. Experimentalmente se observó una buena estabilidad catalítica en la conversión del aceite de canola, en consistencia con otros estudios reportados en la literatura. Con base al contenido de ácidos grasos insaturados, también se esperaba una buena estabilidad en la conversión de lecitina; sin embargo, en las condiciones usadas en este estudio, se propició la formación de sólidos después de procesar 2 g de lecitina, probablemente asociados la fosfatidilcolina y/o a productos de su degradación. Esta rápida desactivación del catalizador durante la conversión de lecitina sugiere claramente la pertinencia de un pretratamiento térmico para fragmentar al aminoácido y el diglicérido; una vez separada esta mezcla, los diglicéridos podrían ser alimentados al sistema de reacción para generar la gama de productos representativa de los combustibles verdes.
Por otra parte, solo algunos trabajos han documentado efecto de WHSV en la conversión de aceites vegetales. Por ejemplo, Prasad y col., (1986), reportaron que en el rompimiento catalítico de aceite de canola sobre HZSM-5, la conversión se incrementó hasta un 10% al disminuir la WHSV de 4 a 2 h−1, sin encontrar efecto en la selectividad hacia hidrocarburos aromáticos. En otro estudio (Siswanto y col., 2008) se evaluó efecto de la WHSV (15 a 25 h−1) en la conversión de aceite de palma sobre H-MCM41; en este caso, la producción de gasolina se maximizó a WHSV 19.38 h−1. Los resultados de la conversión del aceite de canola en el presente trabajo son, en general, consistentes con la literatura, ya que la conversión se incrementó al disminuir la WHSV de 10 a 0.1 h−1: Para el caso de la lecitina, el efecto de la WHSV en la conversión no puede ser concluyente, ya que puede estar enmascarado por la formación del sólido asociado a la fosfatidilcolina y/o a su descomposición.
De forma importante, en este trabajo se realizó un estudio más detallado del efecto de la WHSV en las diferentes fracciones del producto observadas durante la conversión del aceite de canola. Los resultados incluidos en la sección anterior documentaron que la WHSV tuvo un efecto en el rendimiento de CO y CO2 que, mecanísticamente, son productos que se atribuyen a las rutas de reacción de descarbonilación y descarboxilación, respectivamente. La producción de CO y CO2 está documentada ampliamente para el rompimiento catalítico de triglicéridos (Vonghia y col., 1995; Katikaneni y col., 1996; Ooi y col., 2005; Huber y col., 2007), pero pocos autores han reportado datos del efecto de WHSV en de la producción relativa de CO y CO2. La Tabla 4 incluye algunos datos reportados para la selectividad hacia CO y CO2 en el rompimiento catalítico de aceites vegetales sobre diferentes aluminosilicatos. Los datos reportados por Chen y col. (2010) indican que para valores de WHSV de 1 h−1 la relación CO/CO2 fue cercana a la unidad, en congruencia con los resultados de este trabajo. En los datos reportados por Katikaneni y col. (1996) para una WHSV de 3.6 h−1, el mecanismo de descarboxilación fue favorecido en mayor medida que el mecanismo de descarbonilación debido probablemente al uso de un catalizador microporoso y a la presencia de vapor. Sin embargo, cuando la WHSV disminuyó a 1.8 h−1, ambos mecanismos fueron favorecidos en la misma medida, de forma congruente con la tendencia encontrada en este trabajo. Similarmente, Ooi y col. (2005), reportaron que en experimentos a WHSV de 2.5 h−1 la relación CO/CO2 fue alrededor de 2.5. Esta información sugiere que el mecanismo de descarboxilación es más lento, probablemente debido a que involucra más etapas que el mecanismo de descarbonilación. De ser el caso, es posible explicar el cambio observado en la relación CO/CO2 para los experimentos realizados en este trabajo a WHSV menores a 1 h−1, en función del balance de dos efectos acoplados. Cuando se incrementa el tiempo de contacto entre el aceite y el catalizador (menor WHSV), se favorece el mecanismo más lento (descarboxilación).
Tabla 4 Datos de selectividad hacia CO y CO2 para diferentes catalizadores en reactores continuos, a presión atmosférica y temperatura entre 400 y 450 C.
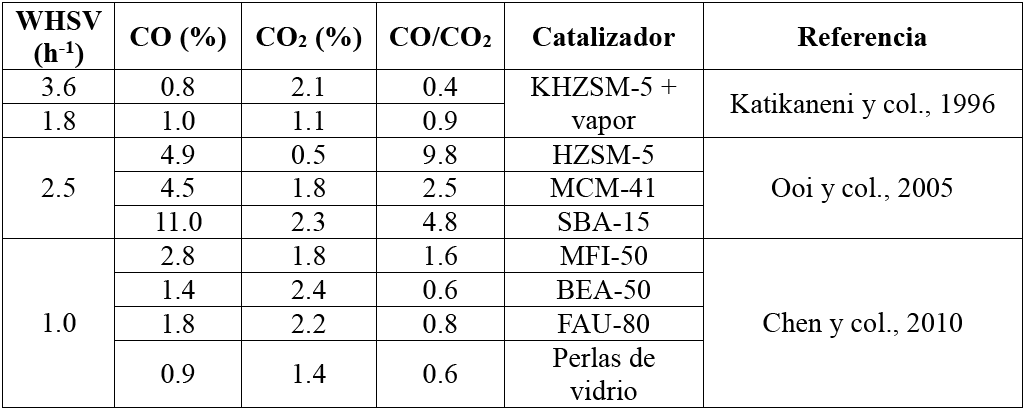
Por otra parte, al disminuir la WHSV la masa de aceite transformada por unidad de tiempo también disminuye, y por tanto, la cantidad de hidrógeno disponible en la superficie del catalizador también disminuye. Esta situación limita notoriamente la ocurrencia del mecanismo de descarbonilación, que requiere de hidrógeno, a pesar de ser más rápido que el mecanismo de descarboxilación.
4.2 Mecanismos
Una observación importante es que en las condiciones experimentales utilizadas en este trabajo, los productos de la conversión de aceite de canola y lecitina son similares a los reportados en la literatura para las zeolitas microporosas, consistente con la presencia de sitios ácidos Brönsted y Lewis. Sin embargo, una diferencia importante con respecto al sistema con catalizador microporoso es que en este trabajo no se detectó la producción de aromáticos como BTX. Estos productos se han asociado a procesos de selectividad de forma característicos de la microporosidad de las zeolitas y, consecuentemente, no eran de esperarse en un material mesoporoso como el H-MCM41 utilizado en este trabajo. Con base a lo anterior, se puede postular que la conversión de aceite de canola y lecitina sobre H-MCM41, es similar al proceso de conversión que ocurre sobre una zeolita que no presenta selectividad de forma. En consecuencia, y tomando en cuenta la distribución de productos observadas en este trabajo, la conversión del aceite de canola y de la lecitina sobre H-MCM41 se puede describir con un modelo similar al propuesto por Benson y col., (2009). Específicamente, la Figura 9 esquematiza las principales rutas de reacción que se proponen en este trabajo para explicar la conversión de aceite de canola y de lecitina en presencia de H-MCM41. Se postula que el rompimiento inicial de la molécula de triglicérido puede ocurrir a través de cuatro procesos principales que ocurren paralelamente:
a) El rompimiento térmico de la molécula de glicerol en el triglicérido
b) La desoxigenación sobre la superficie del catalizador.
c) La adsorción de la molécula insaturada sobre un sitio ácido Brönsted justo en la insaturación (doble enlace) de los ácidos grasos, para dar inicio a los procesos característicos de cracking catalítico.
d) Condensación para formar coque de forma directa.
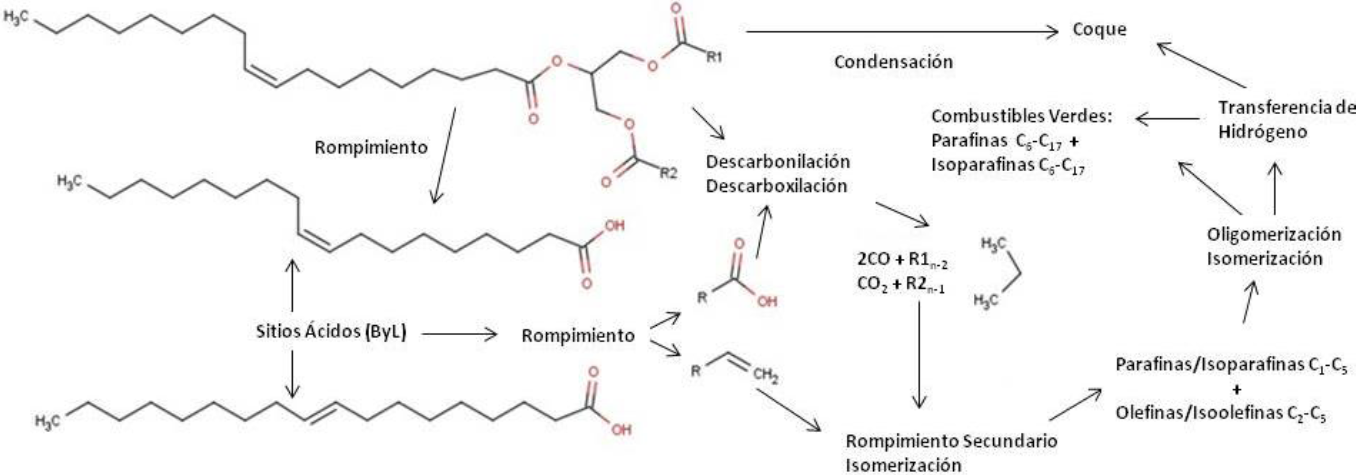
Fig. 9 Mecanismo para el rompimiento catalítico de aceite de canola sobre H-MCM41 propuesto en este trabajo.
Las evidencias experimentales en las que se sustenta la propuesta anterior son las siguientes.
a) El rompimiento térmico del esqueleto de glicerol genera ácidos grasos libres, los cuales pueden participar posteriormente en las etapas de desoxigenación y rompimiento, hasta generar olefinas de bajo peso molecular, CO y CO2. Esta etapa fue evidenciada por la liberación de los ácidos grasos identificados en los experimentos de conversión de aceite de canola a WHSV de 10 y 2.5 h−1. Ciertamente, los ácidos grasos no se detectaron en los experimentos a WHSV menor a 2.5 h−1, pero se puede deber a que estas condiciones experimentales los ácidos grasos fueron consumidos en los procesos de rompimiento catalítico.
b) Las reacciones de desoxigenación liberan CO, CO2, propano, e incluyen olefinas de longitudes de cadena entre C14-C17 que reaccionan rápidamente. En este trabajo, se detectó la presencia de moléculas desoxigenadas C16 y C17 que se incluyó como la "fracción pesada" en la mezcla de productos líquidos. En la Figura 8, la "fracción pesada" se observa en todos los experimentos, independientemente del valor de WHSV.
c) La adsorción del éster de ácido graso en un sitio ácido del catalizador en el carbono donde se presenta una insaturación. Esta etapa se postuló debido a la presencia de mezclas racémicas en el análisis de los productos de reacción. Es importante hacer notar que diversos trabajos en la literatura han documentado los procesos de iniciación de rompimiento catalítico de parafinas mediante la adición de pequeñas cantidades de olefinas en la alimentación, las cuales, al adsorberse sobre un sitio ácido tipo Brönsted, generan entonces los procesos típicos de la química de carbocatión que explican el rompimiento catalítico (Sanchez-Castillo y col., 2002, 2003, 2005). Por otra parte, la formación de mezclas racémicas se ha observado anteriormente en los productos de reacción de diversas olefinas sobre sitos ácidos (Pater y col., 1999). En el caso de este trabajo, las moléculas de triglicéridos alimentadas al sistema de reacción sólo contenían la conformación Z del éster de ácido graso. Sin embargo, las moléculas de ácidos grasos libres observadas en los productos de la conversión del aceite de canola incluyó una mezcla racémica de ácido Z-octadecenoico y del ácido E-octadecenoico, que se debió formar durante la reacción. De esta forma, se postuló que las moléculas de ácido Z-octadenoico se adsorbieron en sitios ácidos Brönsted en la doble ligadura del carbono 9. Debido a la reversibilidad del proceso de adsorción, la molécula adsorbida puede tomar dos rutas. Una de las rutas es desorberse para generar la conformación E del ácido octadecenoico. La otra ruta, inicia el complejo proceso de reacciones asociadas al rompimiento catalítico (oligomerización/escisión β, isomerización y transferencia de hidruro) que, en conjunto, puede dar lugar a una gran variedad de parafinas, olefinas y coque.
d) En las condiciones y tiempos de operación usados para este estudio, no se evidenció la formación de coque, aunque es evidente que la familia de reacciones implicadas en el rompimiento catalítico conducen eventualmente a la formación de coque.
Se debe hacer notar que las parafinas y olefinas generadas por los procesos (a) y (b) participan simultáneamente en la red de familias de reacción indicadas en (c). De esta forma, y de acuerdo a los resultados experimentales de este trabajo, la distribución de los productos de reacción puede ser muy amplia, incluyendo parafinas y olefinas, lineales y con arborescencias, de cadena más corta, particularmente en el rango C6 -C17. El sistema de reacción antes descrito también puede dar lugar a parafinas y olefinas de cadena más larga que puede ser favorables precursores de coque. Por otra parte, se reitera que en las condiciones experimentales de este trabajo no se observó la producción de BTX. En caso requerido para describir la conversión sobre catalizadores microporosos, al modelo propuesto en este trabajo se deberá incluir una familia de reacciones de ciclización para explicar la formación de BTX.
Con respecto al rompimiento catalítico de un fosfolípido, se postula que la parte más reactiva de la molécula, la unión de glicerol con los ácidos grasos y el aminoácido, se fragmenta inicialmente por efecto de la temperatura sobre la superficie del catalizador, generando un aminoácido y un diglicérido. Después, el aminoácido liberado contribuye a la formación del sólido carbonoso que bloquea la superficie del catalizador, y por lo tanto, el acceso a los sitios activos. Sin embargo, previo a esta condición, es posible generar la mezcla de hidrocarburos líquidos similar a la gasolina y al diésel, como la que se observa en el rompimiento catalítico del aceite de canola. Se enfatiza que la propuesta está soportada en el hecho de que el sistema de reacción se validó para la conversión del aceite de canola en condiciones de reacción equivalentes, generando productos líquidos y gaseosos muy similares; por esto, se propone que la química del proceso es análoga entre ambas materias primas. Una estrategia posterior para el uso de fosfolípidos como materia prima para la producción de combustibles verdes, debe implicar una etapa de pretratamiento térmico. Este pretratamiento fragmentará al aminoácido y el diglicérido, y una vez separada esta mezcla, los diglicéridos generados podrán ser alimentados al sistema de reacción, esperando una gama de productos similar a la obtenida para el aceite de canola, sin el problema de desactivación observado a la fecha.
Finalmente, en el mecanismo general de rompimiento catalítico para alcanzar una determinada fracción de productos, es importante hacer notar que la ocurrencia preferencial de una determinada familia o secuencia de reacciones está determinada por la naturaleza de la alimentación, la configuración del sitio activo y por las condiciones de operación, principalmente la composición, la temperatura y la velocidad espacial. La complejidad y el número de reacciones involucradas en este tipo de sistemas no favorecen la descripción detallada de procesos elementales, sino más bien el desarrollo de modelos de lumping de reacciones. A pesar de las ventajas prácticas de este tipo de modelos para el diseño y simulación de equipos y procesos, limita el entendimiento necesario para establecer mecanismos más precisos y para guiar el diseño de nuevos o mejores catalizadores. Un enfoque paralelo para sí permite un entendimiento a nivel fundamental de los procesos involucrado implica el uso de modelos microcinéticos, desarrollados para describir el rompimiento de hidrocarburos de moléculas modelo en sitios ácidos Brönsted (Sanchez Castillo et al., 2002, 2003, 2005). Estos modelos podrán facilitar a futuro la selección cualitativa de las condiciones de operación en las cuales ocurre preferentemente cada familia de reacción.
Conclusiones
En este trabajo se construyó un reactor tubular de lecho fijo para la evaluar el potencial de moléculas derivadas de biomasa en productos en el rango de combustibles verdes, gasolina y/o diésel. El sistema de reacción se validó mediante la conversión de aceite de canola sobre H-MCM41 a presión atmosférica y 400 C. Los resultados, consistentes con la literatura, indican una distribución de productos con una favorable proporción de gasolina y diesel verdes. Se documentó que la proporción relativa de estos productos es función de la WHSV. A partir de los resultados experimentarles se propuso una variante en el mecanismo de conversión del aceite de canola sobre un catalizador ácido mesoporo (H-MCM41), identificando las etapas críticas en la conversión de este tipo de materia primas, y documenta específicamente la iniciación por adsorción de la cadena en la posición del doble enlace sobre un sitio Brönsted, a las condiciones de reacción utilizadas. El mecanismo sugerido implica un rol preferente de los sitios Brönsted sobre los sitios Lewis del catalizador; esta observación deberá guiar el desarrollo de catalizadores con la acidez apropiada para asegurar la estabilidad y selectividad del proceso.
El sistema de reacción también se utilizó para evaluar, bajo las mismas condiciones experimentales usada para el aceite de canola, la conversión de lecitina en gasolina verde, como un sistema modelo para determinar el posible uso de fosfolípidos presentes en la biomasa residual de diversos tipos de industrias, como materia prima alternativa en la síntesis de combustibles verdes. Los productos de reacción identificados al inicio de esta reacción indican que la distribución de productos es similar a la observada previamente para el aceite de canola, determinando así un favorable potencial para generar combustibles verdes. De esta forma, se establecen las bases para guiar el desarrollo de procesos que aprovechan como materia prima un subproducto importante en la industria de la fermentación en un producto de alto valor agregado, incluyendo la pertinencia de los pretratamientos de la materia prima para mejorar la estabilidad del proceso.

