











 nueva página del texto (beta)
nueva página del texto (beta) Inglés (pdf)
Inglés (pdf)
 Artículo en XML
Artículo en XML Referencias del artículo
Referencias del artículo
 Enviar artículo por email
Enviar artículo por email Citado por SciELO
Citado por SciELO  Similares en
SciELO
Similares en
SciELO 
 Permalink
PermalinkINTRODUCTION
Progressive Familial Intrahepatic Cholestasis type 2 (PFIC2) is an autosomal recessive cholestatic liver disease resulting from mutations in the ABCB11 gene, which encodes the bile salt export protein (BSEP), essential for the proper secretion of bile. Patients with PFIC2 typically present in infancy with cholestasis, liver fibrosis, hepatomegaly and severe pruritus and are at risk for developing end-stage liver failure and hepatocellular carcinoma (HCC) within the first decade of life.1 , 2 The prognosis of PFIC2 varies, depending on the severity of the disease and the outcome of treatment. Ursodeoxycholic acid therapy (UDCA) and biliary diversion (PBED) are the only treatments currently available however with mixed outcomes. 3 , 4 Liver transplantation is ultimately necessary in patients who develop progressive liver fibrosis, cirrhosis and end-stage liver disease or as a treatment for intractable pruritus or severe cholestasis.5
We present two siblings with a mild form of PFIC2 that were found to be compound heterozygotes for two rare variants in the ABCB11 gene.
Case 1
8-year-old female who presented at six months of age after having severe bruising at a vaccination site. The vaccine was given after she had an intercurrent infection that was treated with azithromycin. She was noted to be jaundiced and had mild hepatomegaly. The patient had an unremarkable family history. Blood work revealed normal gamma-glutamyl transferase (GGT) cholestasis and mildly elevated international normalized ration (INR) completely corrected with vitamin K administration. During the hospital admission the direct bilirubin increased to 8.2 mg/dL out of a total of 10.4 mg/dL. Abdominal ultrasound showed increased hepatic echogenicity with a contracted gallbladder. A liver biopsy demonstrated hepatocyte swelling with pseudorosette formation and multinucleation, accompanied by hepatocellular and canalicular cholestasis (Figure 1A). Bile ducts were difficult to discern in about half of the portal tracts. Interestingly, many portal tracts lacked intralobular bile ducts by CK7 immunohistochemisty but CK7 was aberrantly expressed in the lobular parenchyma (Figure IB). There was mild portal and perisinusoidal fibrosis seen by trichrome stain. Staining for MDR3 showed retained canalicular staining (Figure 1C). Transmission electron micrographs (EM) showed distended canaliculi containing granular bile and loss of microvilli (Figure ID). The patient’s normal GGT cholestasis resolved by 9 months of age. Genetic testing at 12 months of age revealed a three variants in the ABCB11 gene: c.203G>A, p.(C68Y), c.1331T>C (p.V444A), and c.2495G>A, p.(R832H). The patient continues to follow up regularly in the past 8 years and she remains asymptomatic and is thriving. Routine blood work, including fat- soluble vitamin levels, is unremarkable. Surveillance for hepatocellular carcinoma (with yearly ultrasounds and alpha-feto-protein level) has been negative.
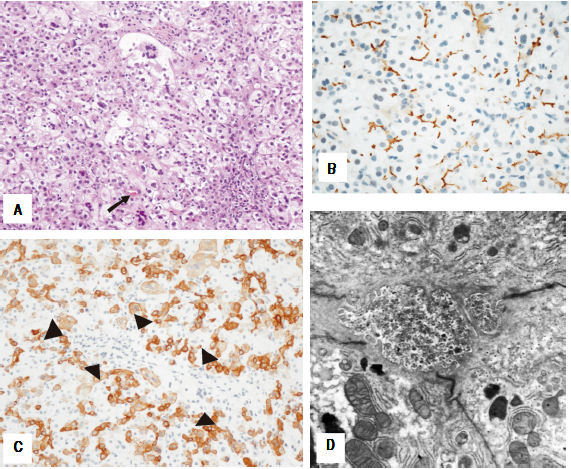
Figure 1 Case 1. Histologic features of the liver needle core biopsy. A(200X H&E): Hepatic lobules show hepatocellular swelling, pseudorosettes and multinucleation with canalicular cholestasis (arrow). B(200X CK7): Immunohistochemistry with cytokeratin 7 shows absence of an interlobular bile duct in the portal tract (arrowheads) in association with aberrant staining of the hepatocytes. C(400X BSEP): BSEP immunohistochemistry shows retained canalicular staining of hepatocyte cell membranes (C-400X). Transmission electron microscopy shows canaliculi distended by amorphous granular bile and loss of microvilli. Mitochondria are unremarkable (D- 11000X).
Case 2
This was the younger male sibling of case 1. A 5-year male who was diagnosed with PFIC2 when cord blood genetic testing revealed that the patient was a compound heterozygote for the c.203G>A and c.2495G> A variant. Further testing of the parents revealed that the c.203G>A, p.C68Y mutation was inherited from the father and the c.2495G>A p.R832H mutation was inherited from the mother. Genetic testing further identified that he had a third polymorphism, a heterozygosity for the sequence variant, c.1331 T>C. Clinically he had mild neonatal jaundice that resolved within the first week of life. At the age of 2 months, he was noted to have mild coagulopathy with elevated INR of 1.8 that resolved with parenteral vitamin K administration followed by oral fat soluble vitamins supplementation. At the age of four months he had pruritus and blood work was notable for elevated serum bile acids with normal aminotransferases, GGT and bilirubin. He was treated with hydroxyzine and cholestyramine, which alleviated the pruritus with concomitant decrease in serum bile acids level. At the age of 12 months cholestyramine and hydroxyzine were discontinued successfully without sequential pruritus. He also stopped oral fat-soluble vitamins supplementation at the age of 12 months and has had normal fat-soluble vitamin levels since. He is thriving well and remains asymptomatic. He continues to have surveillance liver ultrasounds and alpha-feto-protein levels, all of which have been unremarkable.
DISCUSSION
PFIC2 is caused by the inhibition of BSEP, which is responsible for the transport of bile acids from hepatocytes into the canaliculus.6 The inability of bile to enter the canaliculus leads to a buildup within hepatocytes, and damage to the liver. Histopathology reveals hepatocellular and canalicular cholestasis with bile plugs and progressive centrizonal/sinusoidal as well as portal fibrosis. Features of neonatal hepatitis-like changes (inflammation, giant cells, and necrosis) are variable.7 Immunohistochemical staining for BSEP can potentially distinguishes PFIC type 2 from type 1, as patients with PFIC2 have BSEP deficiency while patients with PFIC1 do not. However, positive canalicular staining does not rule out PFIC2 as the channel protein maybe present at the canalicular surface, causing immunohistochemical reactivity, but the protein may have decreased or loss of function.7 The patient in case 1 had portal tracts that lacked intralobular bile ducts by CK7 immunohistochemisty, which is not a typical finding of PFIC2 however the rest of the histologic findings were typical. EM findings show canalicular dilation, microvilli loss, and mild mitochondrial abnormalities. The canalicular bile in PFIC2 patients is non-specific in appearance, unlike the characteristic coarsely particulate "Byler Bile" of PFIC1. Patients with PFIC2 typically present with jaundice, severe pruritus, and poor weight gain. Most patients ultimately develop end-stage liver failure and some require transplantation.
In the case of the siblings we describe above, both patients exhibited an extremely mild form of PFIC2. The elder sibling developed fulminant hepatitis likely as a result of either a viral infection or the use of azithromycin. The younger sibling had pruritus, fat-soluble vitamins deficiency and elevated serum bile acids that have resolved by the age of 12 months. They have both been asymptomatic since, thriving well and do not have fat-soluble deficiencies or any other laboratory abnormality past infancy. It has been shown that there are genotype-phenotype correlations in some mutations8 and better prognosis and response to UDCA or PBED in a few children with missense mutations9 but we are not aware of any other cases of PFIC2 that have followed such a benign clinical course.
Genetic testing of both patients revealed identical rare genetic abnormalities. There are over 80 mutations in the ABC 11 gene described for patients with PFIC2.6 , 10 Genetic testing of both siblings, revealed that they had triple BSEP polymorphisms. Two of the missense variants identified to be C68Y and R832H. Genetic sequencing were also performed in the parents, showing that they each carried one of the mutant alleles, supporting the existence of a compound heterozygous state in both siblings. The first missense mutation, c.203G>A, p.(C68Y) (NM_003742.2) is caused by the substitution of tyrosine for cysteine at amino acid 68 in exon 5. The amino acid is moderately conserved down to chicken. The Grantham distance reveals a large physicochemical difference, and multiple software algorithms predict this variant to be damaging to protein function. This variant has never been reported in population databases to date.
The second missense mutation, c.2495G>A, p.(R832H) is caused by the substitution of histidine for arginine at amino acid 832 in exon. (21) This amino acid is highly conserved, including C. elegans. The Grantham distance identifies this amino acid substitution to be conservative; however, multiple mutation prediction software algorithms classify this variant as damaging to the function of BSEP. This variant has only been identified once in a population database as a heterozygote.11 By strict clinical variant classification guidelines, both c.203G>A and c.2495G>A are considered variants of uncertain significance.
It is of interest that the patients had a common polymorphism, c.1331T>C (p.V444A), which is associated with drug induced liver disease. It is possible that the azithromycin caused decompensation of the underlying liver disease of the patient in case 1.13
Benign recurrent intrahepatic cholestasis (BRIC) is a rare hepatic disease that is associated with PFIC. There are two known subtypes, BRIC1 and BRIC2 that are genetically distinct. BRIC2, caused by mutations in the ABCB11 gene, results in inhibition of the function of BSEP, but differs from PFIC2 in the severity of symptoms and hepatic damage. Patients with BRIC2, present with cholestatic episodes that can range from weeks to months. Although symptoms of BRIC2 can occur in childhood, patients with BRIC2 typically present in adulthood.10 Histopathology of the liver does not show inflammatory intrahepatic cholestasis or liver fibrosis and laboratory tests reveal insignificant hepatocellular injury.14
We consider our patients to have PFIC2 and not BRIC as they both presented in infancy, did not have an episodic course, and the elder patient had biopsy findings compatible with PFIC2 including fibrosis. Both siblings displayed an extremely mild form of PFIC2 with symptoms prominent in infancy that have since resolved. An immunohistochemical stain revealed the presence of BSEP at the canalicular surface; however, based on the genetic alterations (missense mutations) found in the patients, described in detail below, we feel this is likely a non-function or poorly functioning protein. Tumor surveillance with liver ultrasound and alpha-feto-protein is usually recommended every 6 months for patients with PFIC2 though it seems that as our patients have a mild form of PFIC2, with no sign of cirrhosis, it is unclear their risk for developing HCC.

