











 nova página do texto(beta)
nova página do texto(beta) Espanhol (pdf)
Espanhol (pdf)
 Artigo em XML
Artigo em XML Referências do artigo
Referências do artigo
 Enviar este artigo por email
Enviar este artigo por email Citado por SciELO
Citado por SciELO  Similares em
SciELO
Similares em
SciELO 
 Permalink
PermalinkIntroducción
Los linfomas cutáneos primarios son un grupo heterogéneo de neoplasias de células T y B que se presentan en la piel sin evidencia de involucro extracutáneo al momento del diagnóstico. El término linfoma cutáneo de células T (LCCT) describe un conjunto de neoplasias alojadas en la piel que muestran algunas diferencias en la presentación clínica, la histología, el inmunofenotipo y el pronóstico. El LCCT representa del 75-80% de los linfomas cutáneos primarios. La micosis fungoide y las neoplasias linfoproliferativas cutáneas CD30+ constituyen el 90% de los linfomas cutáneos de células T1-3.
El linfoma cutáneo de células T paniculítico (LCCTP) es una neoplasia primaria, muy infrecuente, que representa menos del 1% de los linfomas cutáneos de células T2-4. Es un linfoma de células T periféricas derivado de células T citotóxicas maduras que frecuentemente imitan una paniculitis. Originalmente fue descrito como un linfoma agresivo de células T citotóxicas que predominantemente infiltra el tejido celular subcutáneo y, en ocasiones, puede estar asociado con un síndrome hemofagocítico. Los pacientes clínicamente expresan nódulos subcutáneos o placas, que en la histología corresponden a infiltrados localizados en el tejido celular subcutáneo, generalmente respetando la epidermis suprayacente.
En el 2001, la Organización Mundial de la Salud incluyó en la clasificación de los linfomas cutáneos de células T paniculíticos casos con fenotipo de células T αβ y células T γδ. Los estudios muestran diferencias clínicas, histológicas e inmunofenotípicas entre los casos de LCCTP con estos fenotipos, lo que sugiere que son diferentes entidades, donde el fenotipo γδ presenta un comportamiento muy agresivo.
El diagnóstico de linfoma de células T paniculítico se realiza mediante una biopsia de piel que incluya tejido celular subcutáneo. La histopatología se caracteriza por presentar un infiltrado constituido por linfocitos atípicos que característicamente rodean a los adipocitos y expresan CD3, CD8, receptores de células T αβ y proteínas granulares citotóxicas (granzima B, antígeno intracelular de célula T y perforina). El diagnóstico diferencial del linfoma de células T paniculítico incluye lesiones benignas y malignas que presenten infiltrados en el tejido celular subcutáneo. El tratamiento para este tipo de linfoma depende del fenotipo. Los pacientes que presentan el fenotipo αβ generalmente cursan con una enfermedad indolente confinada al tejido celular subcutáneo, con un pronóstico favorable y una tasa de sobrevida del 82% a 5 años. Los pacientes con fenotipo γδ presentan un comportamiento muy agresivo, similar a otros linfomas de células T γδ, como el hepatoesplénico5, con una tasa de sobrevida del 11% a 5 años, por lo que estos pacientes deben recibir tratamiento agresivo con quimioterapia y considerar la necesidad de un trasplante de células hematopoyéticas6.
En este trabajo se describe el caso de un paciente de sexo masculino con linfoma cutáneo de células T paniculítico que clínicamente mostró características atípicas.
Caso clínico
Se trata de un paciente de sexo masculino de 12 años, madre finada por posible trastorno hematológico a los 26 años; no se reportaron otros antecedentes de importancia. El paciente acudió con dermatosis pruriginosa localizada en el tórax de 2 años de evolución, con diagnóstico de dermatitis atópica. Recibió tratamiento con esteroides, antibióticos y antimicóticos tópicos, sin mejoría. Presentó una evolución tórpida y, 7 meses antes de su llegada, mostró progresión a cara y extremidades, motivo por el cual fue trasladado a nuestro servicio. En la exploración física, se detectó dermatosis que afectaba los cuatro segmentos corporales; la piel cabelluda se encontró constituida por grandes placas costrosas y necróticas; en la cara se detectaron placas eritematocostrosas con edema de párpados que limitaban la apertura del ojo derecho (Figura 1A). En miembros inferiores se encontraron placas ulceradas con costras hematonecróticas, y en rodillas, vesículas y ampollas tensas serosas, además de cicatrices de aspecto atrófico (Figura 1B, 1C y 1D). También se detectó edema de miembros inferiores, con disminución de la fuerza muscular (2/5). Los resultados de laboratorios mostraron anemia, leucopenia, linfopenia y neutropenia, además de discreta elevación de enzimas hepáticas y proteína C reactiva positiva. La propuesta diagnóstica fue linfoma, una vasculitis leucocitoclástica y una paniculitis lúpica, por lo cual se hospitalizó para iniciar con el protocolo de estudio. Se le realizó biopsia incisional que reportó necrosis epidérmica con desprendimiento dermoepidérmico y formación de vesículas, sin epidermotropismo, y neoplasia linfoide en tejido celular subcutáneo con patrón panlobular que comprometía todos los anexos, rodeándolos, sin vasculitis. Esta neoplasia estaba constituida por células con núcleos hendidos e, hipercromáticos con citoplasma escaso. Se identificaron cuerpos apoptóticos y múltiples mitosis atípicas (Figura 2). Los linfocitos atípicos expresaban CD3, CD8, CD4 y CD7 y fueron negativos para CD30 y CD19 (Figura 3). No fue posible llevar a cabo el rearreglo genético para el receptor de células T debido a las limitaciones de nuestro medio hospitalario. Con estos datos, el paciente fue evaluado por el servicio de oncología, donde se le realizaron estudios de extensión, radiografía de tórax que no demostró masa mediastinal, ultrasonido de abdomen que reportó hepatoesplenomegalia, tomografía computarizada que mostró ganglios cervicales, axilares e inguinales, y aspirado de medula ósea, que fue negativo para células neoplásicas. Con base en estos datos previos se confirmó el diagnóstico de linfoma no Hodgkin de células T paniculítico estadio IIIA y se descartó el síndrome hemofagocítico. Posteriormente se inició tratamiento con ciclofosfamida, doxorrubicina, vincristina y prednisona.

Figura 1 A: dermatosis diseminada constituida por grandes placas costrosas de aspecto psoriasiforme; en cara se observan placas eritematocostrosas con edema de párpados que limita la pertura del ojo derecho. B: miembros inferiores con placas ulceradas con costras hematonecróticas; en rodillas, vesículas y ampollas tensas serosas, cicatrices de aspecto atrófico y edema de miembros inferiores. C-D: acercamientos de las lesiones. Se muestra el polimorfismo y resalta la presencia de ampollas y la necrosis cutánea.
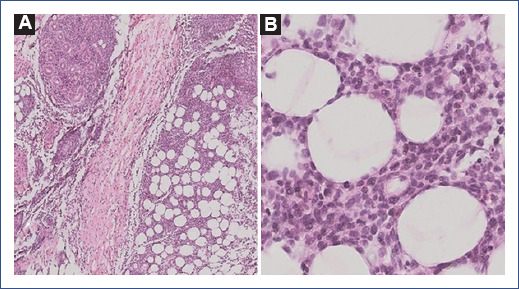
Figura 2 A: tinción hematoxilina-eosina, 4x. Se observa un denso infiltrado de predominio linfocítico periglandular a nivel lobulillar en el tejido adiposo. B: tinción hematoxilina-eosina, 10x. En este acercamiento se aprecian las características de este infiltrado atípico, con pleomorfismo nuclear; se observa la disposición de estas células linfoides en la periferia del adipocito y algunas mitosis atípicas.

Discusión
El LCCTP es un linfoma de células T periféricas que representa menos del 1% de los subtipos de linfoma no Hodking. La incidencia exacta y su variación según la región no se ha descrito apropiadamente. Se presenta en pacientes con una media de edad de 36 años, aunque aproximadamente el 20% de los pacientes son menores de 20 años. Los casos pediátricos son raros, con solo algunos reportes en la literatura7. Existe un predominio por el sexo femenino (relación 0.5). Más del 20% de los pacientes presentan LCCTP asociado con una entidad autoinmune como lupus eritematoso sistémico, artritis idiopática juvenil, diabetes mellitus tipo 1 o síndrome de Sjögren1.
Cuando en algunos pacientes existe regresión espontánea o un prologado y latente curso clínico, la mayoría de los casos tienden a ser agresivos. El LCCTP es un subtipo de linfoma cutáneo caracterizado por una infiltración de células T neoplásicas CD8+8. Puede convertirse en un reto diagnóstico debido al polimorfismo de su expresión clínica, como en el presente caso. Los pacientes con LCCTP presentan una dermatosis en brazos, piernas y tronco constituida por nódulos subcutáneos no dolorosos o placas induradas que varían en su diámetro; en ocasiones pueden desarrollar úlceras, pero esto ocurre con mayor frecuencia en las variantes más agresivas9. Cabe destacar que no encontramos ningún reporte en la literatura de esta variante con la expresión clínica vesículoampollosa. Aproximadamente el 75% de los pacientes con LCCTP tienen un compromiso multifocal en piel6. Existen reportes de afección en cara y cuello en la población pediátrica, como en el paciente del presente caso10. Los síntomas B y las citopenias son comunes en ambos fenotipos. Con respecto a los síntomas sistémicos, la fiebre afecta al 50% de los pacientes; ocasionalmente puede estar acompañada de otros síntomas B, como sudoración nocturna o pérdida de peso4. En una serie de 16 casos de López-Lerma et al., el 25% de los pacientes contaban con historia de patologías autoinmunes; como en otros estudios, la entidad autoinmune más frecuente fue lupus eritematoso8. Huppmann et al. reportaron una asociación del 17% con el síndrome hemofagocítico9.
Histológicamente, el LCCTP se caracteriza por la presencia de un infiltrado de células neoplásicas y linfocitos en el tejido celular subcutáneo11. En los casos de células T γδ, puede existir compromiso epidérmico y dérmico12. Los linfocitos muestran apoptosis, cariorexis, áreas focales de necrosis grasa y características atípicas como núcleos hipercrómicos y bordes mal definidos. También pueden observarse histiocitos, células plasmáticas y neutrófilos simulando una paniculitis benigna, además de mitosis dispersas.
Se realizó el diagnóstico diferencial con linfoma tipo hidroa, debido a que esta entidad afecta principalmente a niños y adolescentes y, desde el punto de vista clínico, se distingue por un marcado edema facial junto con una erupción caracterizada por vesículas, pápulas y zonas de necrosis que dejan cicatrices varioliformes en la cara, el dorso de las manos, los antebrazos y las piernas13. Histológicamente, el presente caso comparte con el linfoma tipo hidroa el predominio del infiltrado linfocitario atípico en la dermis y el tejido celular subcutáneo; sin embargo, en los casos de linfoma tipo hidroa, las características angiocéntricas y angiodestructivas son típicas, a diferencia del linfoma T paniculítico, donde los vasos sanguíneos (como en este caso) no se ven afectados14.
El diagnóstico diferencial también debe realizarse con paniculitis lúpica, la cual presenta varias características histológicas superpuestas con LCCTP. Además, se sabe que la paniculitis lúpica ocurre en asociación con LCCTP en aproximadamente el 20% de los casos. Aunque es un rasgo característico de LCCTP, el borde periférico de los lóbulos de adipocitos por linfocitos atípicos pleomórficos a veces puede observarse en la paniculitis lúpica. Además, las células plasmáticas, que a menudo se cree que favorecen la paniculitis lúpica, también pueden estar presentes en LCCTP, lo que aumenta la confusión diagnóstica15. En un estudio se ha informado que los hallazgos histopatológicos que sugieren paniculitis lúpica coexisten con los de LCCTP en la misma muestra de biopsia. Cuando se habla del diagnóstico diferencial entre estos dos padecimientos, debe considerarse que una paniculitis benigna presenta células B CD20- mezcladas con CD3- CD4- y CD8-, mientras que la paniculitis lúpica comúnmente presenta células T CD4+ y no CD8+6,16,17.
Otra entidad que a menudo se confunde con LCCTP es la paniculitis histiocítica citofágica (PHC), que muestra paniculitis lobulillar y proliferación de histiocitos citofágicos de aspecto benigno en la histología. Bader-Meunier et al. comunicaron dos casos de niños de 6 y 16 meses con clínica similar al presente caso, asociado con PHC. Sin embargo, a diferencia del presente caso, estos autores no encontraron linfocitos atípicos en sus pacientes18. Aunque la relación entre LCCTP y PHC es un tema, muchos investigadores comentan que ambas entidades pueden coexistir.
Otro diagnóstico diferencial es la paniculitis lobulillar linfocítica atípica. El inmunofenotipo es fundamental para el diagnóstico. Las células neoplásicas son células T citotóxicas, generalmente CD3+ y CD4-. La variedad αβ es CD8+ y generalmente CD30- y CD56-, mientras que el subtipo γδ es CD8-, CD56+ y CD30+. En nuestro caso, la biopsia mostró características compatibles inmunofenotípicamente con la variedad ab.
Los regímenes basados en quimioterapia a expensas de antraciclinas han sido utilizados para el tratamiento de LCCTP, pero los resultados aún son limitados, por lo que, frecuentemente, la enfermedad es fatal. Algunos grupos han reportado el uso de altas dosis de quimioterapia seguido del trasplante de células troncales hematopoyéticas como tratamiento de rescate exitoso. Sin embargo, será necesario un seguimiento más prolongado para definir el comportamiento de este padecimiento. En muchos casos, especialmente en aquellos que cursan con síndrome hemofagocítico, la presentación es agresiva19, por lo que se necesitan investigar líneas de quimioterapia más efectivas.
Actualmente, el tratamiento para este tipo de linfomas no se ha estandarizado, pero se sugiere iniciar quimioterapia con ciclofosfamida, doxorrubicina, vincristina y prednisona7; bexaroteno es otra opción terapéutica20. El trasplante de células troncales después de la quimioterapia se ha sugerido como tratamiento para casos recurrentes y refractarios de LCCTP con buenos resultados en algunos casos17,21. Dada la afección cutánea extensa, así como la afección sistémica en el caso aquí presentado, se sospechó un comportamiento agresivo, por lo que se decidió iniciar tratamiento con quimioterapia 10 días después del diagnóstico, con una expectativa de sobrevida del 10-20%22.
Para concluir, en este trabajo reportamos un caso de LCCTP, un linfoma cutáneo infrecuente para este grupo de edad23. Cuando un paciente presenta una larga historia de lesiones que recuerdan paniculitis, no responde a tratamientos tópicos y antibioticoterapia, y no se acompaña de síntomas clínicos específicos, debe considerarse el LCCTP como posibilidad diagnóstica. La neutropenia que no responde a antibióticos es una pista; además, es esencial una biopsia que incluya tejido celular subcutáneo para la confirmación del diagnóstico. Adicionalmente, un estudio de inmunohistoquímica debe realizarse para diferenciar LCTTP de otras entidades y guiar el tratamiento. Retrasar el diagnóstico puede empeorar los síntomas y limitar las posibilidades terapéuticas y pronósticas.
Consideramos de gran importancia esta comunicación por la baja incidencia de reportes en la literatura, la edad de presentación, la expresión excepcional y el hecho de haber tenido una presentación tan agresiva en este caso. De igual manera, por el impacto de la histología acompañada de la inmunohistoquímica, que permitieron establecer el diagnóstico. Es posible que esta entidad se encuentre sub-reportada, por lo que invitamos a los especialistas involucrados a comunicar los casos para contar con una mejor experiencia diagnóstica y terapéutica que influya en la sobrevida del paciente.

