











 nueva página del texto (beta)
nueva página del texto (beta) Inglés (pdf)
Inglés (pdf)
 Artículo en XML
Artículo en XML Referencias del artículo
Referencias del artículo
 Enviar artículo por email
Enviar artículo por email Citado por SciELO
Citado por SciELO  Similares en
SciELO
Similares en
SciELO 
 Permalink
PermalinkIntroduction
Fibrodysplasia ossificans progressiva (FOP) is a rare genetic disease, with an estimated incidence of 0.6-1.3 per million individuals1, characterized by progressive heterotopic endochondral ossification and congenital malformations of the great toes2. It is included in the Online Mendelian Inheritance in Man (OMIM) registry under number 1351003.
FOP evolves in flare-ups that produce new areas of ossification, leading to progressive motor disability and death, mainly due to restrictive respiratory complications4. It is an autosomal dominant disease caused by heterozygous mutations in the ACVR1 gene encoding the activin receptor IA (ACVR1/ALK2), a protein in the bone morphogenetic signaling pathway5.
Although several drugs are used to treat this pathology, there is yet to be a treatment with proven clinical efficacy1.
We report the case of a female patient diagnosed with FOP at the age of 3 years. The objective is to inform the case of a rare disease that generated diagnostic difficulties. Knowledge of this entity can avoid unnecessary paraclinical evaluations that can be iatrogenic and favor disease progression.
Clinical case
We describe the case of a 3-year-old female patient with good growth and development, healthy parents, and two healthy siblings. The patient showed congenital bilateral hallux valgus and had a complete immunization scheme. The patient was evaluated for presenting 2-week evolution of multiple painful cervical tumors in both jugular-carotid regions, predominantly on the right side. She showed fever in the first two days of onset of the disease, remaining in apyrexia afterward. No symptoms of intercurrent infection and general compromise were further observed.
Initially, the tumors were of firm-elastic consistency, 3 cm in diameter, and adhered to the muscular planes. They were accompanied by erythema of the overlying skin, which disappeared during evolution, acquiring a stony consistency. Lymphoganglionic examination revealed multiple mobile adenomegalies in jugular-carotid, submaxillary, axillary, inguinal, and abdominal regions; a painful pole of the spleen was palpated. On admission, the sternocleidomastoid and trapezius muscles had a stony consistency, generating painful torticollis (Figure 1).
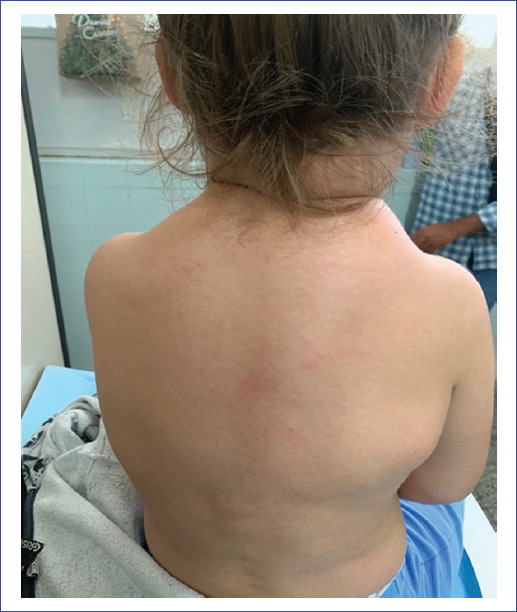
During the 2-week hospital stay, tumors with similar characteristics were detected in other regions related to muscular structures: posterior cervical region, trapezius muscles, right subscapular region, and left paravertebral level (Figure 2). The rest of the physical examination showed no alterations.
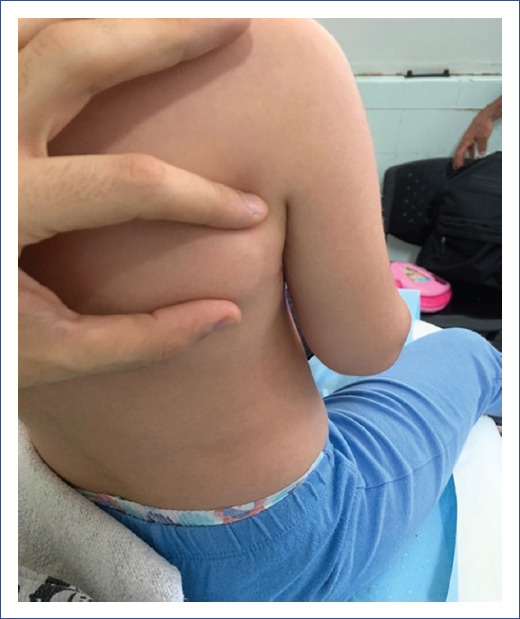
An anatomopathological study and imaging follow-up of the lesions by ultrasound and magnetic resonance imaging (MRI) were performed. Malignancy and ossification were excluded. The rest of the complementary studies showed no alterations.
During outpatient follow-up after discharge, the patient presented partial regression of the tumors that motivated the initial consultation and similar lesions in the pectoral region, right dorsum, and abdominal wall.
Clinical and functional improvement occurred after initiating treatment with prednisolone at 2 mg/kg. However, the symptomatology returned when this dose was reduced. After five months of evolution, the patient presented pain and limitation of movement. The trapezius and sternocleidomastoid muscles, biceps, and triceps of the right upper limb showed a stony consistency, as well as at the biopsy sample extraction sites. Plain radiography of the right upper limb showed calcification of the right biceps brachii muscle (Figure 3).

The presence of muscular calcifications, in addition to the reevaluation in the diagnostic reasoning of congenital hallux valgus-interpreted until now as an isolated morphological alteration-allowed the establishment of the clinical diagnosis of FOP.
The molecular genetic study found ACVR1:c.617G>A (p.Arg206His) in heterozygosis, confirming the clinical diagnosis.
Discussion
This present case is the second case of FOP reported in Uruguay6. It is one of the so-called "ultra-rare" diseases, with an estimated incidence of 0.6-1.3 per million individuals. Diagnosis is clinical and confirmed by molecular studies1.4. No sex predominance has been established.
This autosomal dominant disease is caused by gain-of-function mutations in the ACVR1 gene, located on chromosome 2q23-24. More than 95% of patients have the missense mutation c.617G > A (p.Arg206His), as this patient5.
The ACVR1 gene encodes for the activin receptor type IA, a membrane protein member of the family of intercellular signaling protein receptors called bone morphogenetic proteins (BMP) type I5,7. The p.Arg206His mutation, which consists of changing the amino acid arginine to histidine at position 206, resides in a domain critical for regulating this protein called the GS (glycine-serine) domain. This change causes a gain of function, leading to constitutive or ligand-independent deregulated activation, which explains its dominant expression. This mutation modifies the folding of the protein at the binding site of another protein (FKBP12) with inhibitory activity on ACVR1. The inability to be inactivated by its negative regulator generates constitutive activity of the receptor encoded by ACVR1. The consequent dysregulation of the BMP signaling pathway would lead to aberrant cartilage production from mesenchymal cells8.
Morphological alterations of the distal bones of the limbs are one aspect of the disease. Other recurrent mutations in this gene (p.Gly328Arg, p.Gly328Trp, and p.Gly328Glu) are associated with characteristic limb alterations that may be mistaken for amniotic flange disruptions or type B brachydactyly. The penetrance of gain-of-function mutations in the ACVR1 gene is estimated to be complete, as no cases with these mutations or their clinical expression have been reported5. As in most dominant diseases, wide phenotypic variability can be observed, and other genes and environmental factors likely modulate the presentation of the disease5.
As we propose, most cases are sporadic, arising from a de novo mutation, since none of the parents have elements suggestive of this condition1.
Two clinical elements classically define FOP: malformations of the great toe and progressive heterotopic endochondral ossification in characteristic anatomical patterns. Patients with FOP present as healthy neonates, except for the malformation of the great toe, which is present in all affected individuals, as observed in this case2,4,9.
Initially, this patient presented tumors of firm-elastic consistency in the neck, with adherence to muscular planes. This observation led to other diagnoses, including hemato-oncologic causes, usually one of the most frequent initial approaches10. Initial imaging studies showed no calcifications, which generated diagnostic confusion. Current evidence demonstrates that the development of heterotopic ossification occurs after soft tissue inflammation4,11. Routine biochemical studies are usually normal, as observed in this patient. To date, no specific biomarkers have been identified1.
Hallux valgus in the pediatric age group is usually acquired during infancy and childhood; it is infrequently congenital, as in this case12. Although congenital malformations of the great toes are a constant feature of FOP, their presence is not pathognomonic1,2. In addition, it is possible to observe other alterations that this patient did not present, such as malformation of the thumbs, clinodactyly, tibial osteochondromas, and wide and short femoral necks1,5.
Affected patients develop episodic flare-ups of pain and soft tissue inflammation. Some episodes remit spontaneously, but in most of them, the soft connective tissues are transformed into bone tissue by endochondral ossification, leading progressively to permanent immobility. In addition, minor trauma, such as intramuscular immunizations, dental anesthesia, muscle fatigue, hematomas, falls, and infections, can trigger new flare-ups1,4.
As observed in this patient, heterotopic endochondral ossification progresses following anatomical and temporal patterns that mimic the patterns of bone formation in the normal human embryo. It initially occurs in the dorsal, axial, cranial, and proximal regions of the body and is subsequently seen in the ventral, appendicular, caudal, and distal regions. Some skeletal muscles are unaffected, including the diaphragm, tongue, and extraocular muscles. Cardiac and smooth muscle tissues are also unaffected5,13.
Most patients require assistance with activities of daily living in the early stages. Mandibular involvement is usually followed by significant malnutrition. A frequent cause of death in affected patients is progressive cardiorespiratory failure due to thoracic insufficiency syndrome, which results in the inability of the thorax to achieve normal ventilation. The median age at death is 40 years13,14.
There is currently no definitive medical treatment for FOP. Therefore, management consists of supportive care. Since this is a chronic disease with a significant impact on quality of life, it is recommended that the care of patients with FOP be multidisciplinary1,15.
High doses of glucocorticoids have shown efficacy in the early treatment of flare-ups affecting the major joints, mandible, and submandibular region1,4. They also help prevent flare-ups after trauma and surgery, decreasing the probability of heterotopic ossification in the affected sites1,16. The administration is suggested in the first 24 hours of the outbreak, at a daily dose of 2 mg/kg/day, for no more than 4 days. This patient received corticosteroids and presented a good initial response.
Nonsteroidal anti-inflammatory drugs, cyclooxygenase-2 inhibitors, mast cell stabilizers, and anti-leukotrienes are helpful in pain management17. Other drugs, such as bisphosphonates and imatinib, are not routinely used but may contribute to managing difficult-to-control flare-ups18.
New therapies are being investigated, including palovarotene derived from retinoic acid. This drug reduced the percentage of FOP patients who developed heterotopic ossification and is currently in a phase 3 study19.
Surgical interventions to remove heterotopic bone tissue result in reactive bone regrowth, as observed at the biopsy specimen extraction sites in this patient, and are therefore contraindicated1,10.
Preventive measures aimed at reducing flare-ups and subsequent ossification are essential. Patients with FOP should have prophylactic dental control, excluding mandibular anesthetic blocks. There is consensus on avoiding vaccination and intramuscular administration during flare-ups. Administration of vaccines with diphtheria, pertussis, and tetanus components, which are associated with a higher incidence of flare-ups, is not routinely recommended20. For the same reason, muscle fatigue should be limited, and falls and respiratory infections should be prevented1,10.
In conclusion, FOP is a sporadic disease often underdiagnosed, especially in its early stages. If clinical suspicion exists, early molecular testing is suggested to detect the most frequent mutations of the ACVR1 gene. Current management of FOP consists mainly of symptomatic treatment, maintenance of physical function, and family support.

