











 nueva página del texto (beta)
nueva página del texto (beta) Inglés (pdf)
Inglés (pdf)
 Artículo en XML
Artículo en XML Referencias del artículo
Referencias del artículo
 Enviar artículo por email
Enviar artículo por email Citado por SciELO
Citado por SciELO  Similares en
SciELO
Similares en
SciELO 
 Permalink
PermalinkIntroduction
Neurofibromas are benign peripheral nerve sheath tumors1. Pigmented neurofibroma (PN), also called melanocytic neurofibroma, is a rare subtype of neurofibroma that contains melanin-producing cells and constitutes only 1% of neurofibroma cases2. As other pigmented tumors share the same neuroectodermal origin, the clinical and pathologic diagnosis of these tumors is challenging. The occurrence of hypertrichosis in neurofibromas (including PN) is very rare3. Here, we present the case of PN associated with hypertrichosis in a schoolchild.
Clinical case
We describe the case of an 8-year-2-month-old male patient, native and resident of Oaxaca, Mexico, the only child of non-consanguineous parents. Family history: a father with NF1, normal perinatal data, uncomplicated pregnancy. Birth weight was 3130 g, height 49 cm, and head circumference 35 cm. Currently, the patient presents complete neurodevelopment for his age.
The patient has congenital pseudarthrosis of the left tibia and fibula, requiring osteotomy of the left tibia. In addition, he has right multicystic renal dysplasia, hydronephrosis, secondary arterial hypertension, and neurogenic bladder. Since the patient is a carrier of NF1, he is under multidisciplinary medical follow-up in a tertiary hospital in Mexico City.
At one year of age, his current condition began when he presented a localized dermatosis on the left lower extremity affecting the anterior aspect of the thigh and ipsilateral knee, consisting of a light brown hyperpigmented spot, well-demarcated and smooth in consistency. The lesion was asymptomatic, with slow and progressive growth after the patient's development. An increase in the volume of the area and the appearance of hair follicles (data compatible with hypertrichosis) on the spot's surface were observed at the age of 3 years (Figure 1). Due to the progression of the lesion (increase in volume, number, and diameter of hair follicles), we decided to perform a skin biopsy.
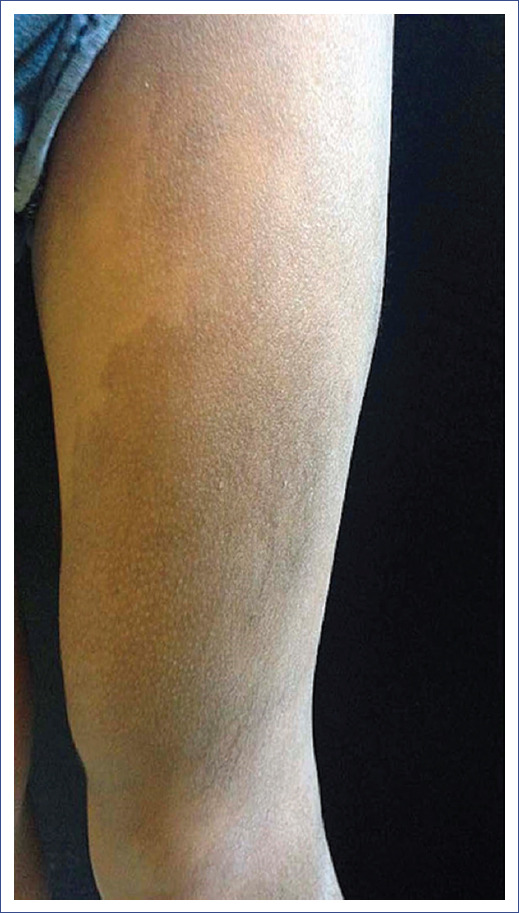
Figure 1 Neoformation with the appearance of a light brown hyperpigmented plaque and well demarcated with hypertrichosis (16 × 9 cm).
Skin biopsy showed an epidermis with stratum corneum in a basket network and flattening of the interpapillary processes. The superficial to mid-reticular dermis showed a proliferation of spindle cells with "italic" S-shaped nuclei intermingled with mast cells and concentrically arranged in a myxoid stroma (Figure 2). In the deep portion of the lesion, melanin deposits were found interspersed between the spindle cells, which stood out with the Fontana-Masson stain (Figure 3A).

Figure 2 A: skin biopsy showing a diffuse spindle cell proliferation affecting the superficial, middle, and deep dermis, as well as adipose tissue. B: superficial portion of the lesion showing spindle cells with "italic" S-shaped nuclei intermixed with mast cells and arranged in a whirling pattern in a lax stroma. C: in the deep portion, more bulky ovoid cells with brown pigment deposits in their cytoplasm are identified.

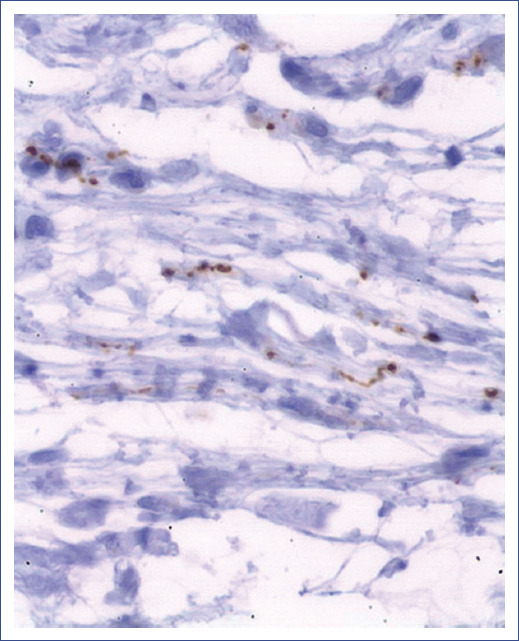
Figure 3 A: Fontana Masson stain positive for melanin in the cytoplasm of ovoid cells in deep dermis and in macrophages. B: CD117 labeling for plasma cells, which intercalate between the dendritic cells of the neurofibroma. C: S100 protein positive (brown) in Schwann cells of neurofibroma. D: Melan-A (in red) positive in melanin-producing spindle cells.
Immunohistochemistry showed CD117-positive mast cells (Figure 3B) and S100-positive spindle cells (Figure 3C). Immunohistochemical markers Melan-A and HMB45 showed some interspersed melanocytes, mainly in the deep portions of the lesion (Figure 3D). Intercalated structures were observed between spindle cells with immunolabeling for neurofilaments (Figure 4). On this basis, the anatomopathological diagnosis of PN was established. The patient continued with his multidisciplinary medical follow-up. Regarding the management of PN, it was decided to monitor the lesion every six months.
Discussion
PN is a cutaneous tumor that affects more females and predominates during the second and third decades of life, although some reports show an age range of 2 to 71 years3,4. These tumors can occur in isolation or be associated with NF1.
PN has been described more frequently in dark-skinned populations: in two series, 86% of patients were from Africa, Asia, or Latin America3,5,6. The most frequent topography of this pathology is the scalp3,5; other locations are the neck, hands, buttocks, thighs, and legs6. Morphologically, the lesions are tumors with no visible pigment in most cases. Occasionally, the lesions are accompanied by hyperpigmented spots with shades ranging from gray to brown and different pigment intensities. In a case series, Fetsch et al. observed that only 16% of the tumors investigated showed macroscopically detectable areas of pigmentation. The lesions had a rubbery consistency with poorly defined borders2. Moreover, a size range from 1 to 50 cm in diameter has been described7; some tumors appear as a single lesion, although two lesions have been identified in the same patient6.
Furthermore, an association between hypertrichosis and neurofibroma is rare, especially in PN2,3,8. This lesion's mechanism for excessive hair growth has not been well established. Some authors consider that localized hypertrichosis may occur due to abnormal signals for follicular papilla elongation, including growth factors (epidermal bone growth factor, vascular endothelial growth factor, platelet-derived growth factor, and bone morphogenic protein) and a prolonged anagen phase of the hair cycle3. Other authors have established the possibility that localized acquired hypertrichosis may occur in areas of friction, trauma, or inflammation, suggesting that local factors may influence these growth mediators and determine the increase in the anagen phase3.
Only five cases of PN associated with hypertrichosis have been published2,3,8. These tumors are usually accompanied by hyperpigmented spots with hypertrichosis on their surface and are lesions > 10 cm. In addition, the lesions appear early in life and progress gradually. Finally, most PNs are part of the clinical spectrum of NF1, with neither sex nor age group predominance.
PN is an exophytic neoformation involving the dermis and subcutaneous cellular tissue, composed of fascicles of spindle or epithelioid cells distributed throughout the tumor, immersed in a stroma with collagen and matrix. Microscopically, PNs present the features of neurofibromas. Most cases show features of diffuse neurofibroma; others resemble the plexiform type, and in other cases, characteristics of both4,6. Occasionally, the spindle cells may be arranged in a storiform pattern7. Most lesions may contain abnormal nerve segments with disorganized Schwann cells and Wagner-Meissner corpuscles6. Other findings include increased mast cells and the occasional presence of randomly distributed multinucleated giant cells in the stroma6.
PN is the only subtype of neurofibroma that contains melanin-producing cells that tend to be located in the deep dermis and subcutaneous cellular tissue; according to Motoi et al., this pattern may be a valuable tool to differentiate PN from other pigmented lesions5. The presence of melanin-producing cells may be due to the origin of melanocytes and Schwann cells, which is from neural crest cells5,9. To date, it has been debated whether the pigmented cells in this tumor are displaced melanocytes or Schwann cells with aberrant melanogenesis6. Detecting microphthalmia transcription factor (MITF) in the nuclei of PN tumor cells further supports the phenotypic relationship between melanocytes and melanin-producing Schwann cells, as this transcription factor is essential for melanocyte development and survival10,11.
Melanin within phagocytes can be identified in different areas of the dermis and subcutaneous cellular tissue when stained with Fontana-Masson and Warthin Starry12. On immunolabeling, the tumor expresses S100 protein and melanin markers4-6: Melan-A, MITF (critical regulatory factor of melanogenesis), and HMB45. At times, these tumors are positive for tyrosinase and C-Met (involved in regulating melanogenesis)5. The characteristic immunohistochemistry of PN suggests that it is a single tumor showing differentiation towards mature melanin production (with mature melanocytes) but with apparently impaired synthesis capacity5,6.
Fetsch et al. proposed some pathological diagnostic criteria for PN in their review: 1) mild histologic appearance to diffuse neurofibroma or a neurofibroma with combined histologic features of plexiform and diffuse; 2) immunolabeling to S-100 protein, HMB-45 antigen, and CD342,6.
PN can be confused with pigmented and hypertrophic tumors or masses. The primary differential diagnoses of this tumor are melanocytic schwannoma13, pigmented dermatofibrosarcoma protuberans (Bednar's tumor)14,15, cellular blue nevus16,17, giant congenital melanocytic nevus with neuroid features18,19, and cutaneous neurocristic hamartoma20-23 (Table 1). To prevent doubts during diagnosis due to the clinical expression of PN, it is always advisable to take a biopsy of the lesion, whose anatomopathological characteristics may define the final diagnosis.
Table 1 Differential diagnoses to be considered in pigmented neurofibroma
| Characteristics | Pigmented neurofibroma | Melanocytic schwannoma 13 | Dermatofibrosarcoma protuberans pigmentosum (Bednar's Tumor)14,15 | Blue cellular nevus 16,17 | Cutaneous neurocristic hamartoma 18,19 | Giant congenital melanocytic nevus with neuroid features 20-23 |
|---|---|---|---|---|---|---|
| Origin | Peripheral nerve sheath: neuro-mesenchymal tissue; perineural, Schwann and melanin-producing cells | Peripheral nerve sheath consisting of Schwann cells | Fibrohistiocytic with dendritic cells | Dendritic melanocytic | Neuro-mesenchyme with fibrogenic, melanogenic and neurosustentacular differentiation | Melanoblasts: nevus cells and Shwann cells |
| Location | Hairy skin, neck and lower extremities | Axial region, nerve roots of the spinal cord, central and autonomic nervous system | Trunk, inguinal region and lower extremities | Buttocks in sacrococcygeal region, hairy skin, face and extremities | Head (in hairy skin) and trunk (thorax and back) | Trunk in bathing suit area and extremities. |
| Clinical manifestations | Hyperpigmented, poorly defined tumors of rubbery consistency | Solid mass with well-demarcated brown and/or black borders | Multinodular pigmented tumor firm to touch/Sclerotic or atrophic plaque | Dark blue neoformation with smooth dome-shaped surface | Focal alopecia Solitary neoformation of nodular appearance | Brown plaque with flat or mammillary surface, well-demarcated borders, hypertrichosis |
| Accompanying symptoms | Asymptomatic | Radicular pain, dysesthesias and progressive sensory-motor involvement. | Asymptomatic | Small painless lesions, large ones painful and ulcerating | Asymptomatic | Asymptomatic occasional pruritus |
| Cell phenotype | Spindle or epithelioid cells | Elongated cells some with epideloid appearance | Mesenchymal spindle cells | Pigmented dendritic melanocytes mixed with epitheloid cells | Nevomelanocytes, schwann cells, pigmented dendritic cells, spindle cells and fibroblasts | Neuroid and Schwan elements |
| Other histological data | Wagner-Meissner bodies, Schwann cells and mast cells | Psammoma bodies (40-50%) Laminated calcifications Mature adipocytes | Pigmented dendritic cells Verocay bodies | Cell islets may form digitate projections or bulbous expansion (dumbbell pattern) | Tactoid bodies Decrease in hair follicles | Elongated or thin melanocytes (type C) with fibrillar collagen structures resembling Meissner's corpuscles or Verocay's bodies |
| S-100 | Positive | Positive | Negative | Positive | Positive | Positive |
| HMB45 and/or Melan-A | Positive | Positive | Negative | Positive | Positive | Positive |
| Other immunomarkers | MIFT, CD34 | Vimentin, Leu7 | CD34+, SMA, CD117, vimentin | SOX10, MART-1 | CD34 and NSE | |
| Associations | Neurofibromatosis 1 | Carney complex Recurrence, malignancy (10%) and metastasis capacity | Intermediate malignancy, high risk of recurrence | Possible transformation to malignant melanoma | Spina bifida, neurocutaneous melanosis and potential for malignant transformation |
No standardized treatment for PN has been established. In some situations, partial resection of the tumor is necessary due to extra weight, limitations in daily activities, and cosmetic improvement. Surveillance is suggested, as some cases of PN present recurrence. No malignant transformation has been shown in any of these tumors, and follow-up data are insufficient to comment specifically on this issue4,7. Finally, there is no treatment to prevent the appearance of PN.
Most of the time, PN appears in individuals with a confirmed diagnosis of neurofibromatosis; therefore, patients already have a multidisciplinary medical follow-up of their genodermatosis and only need clinical observation of the PN. Surveillance or surgical management will be chosen depending on the tumor's evolution. When a PN appears spontaneously, other cutaneous signs of NF1 (more than six café-au-lait spots, axillary or inguinal ephelides, and more neurofibromas)24 should be sought. Patients should be referred to ophthalmology, neurology, orthopedics, cardiology, gastroenterology specialties, and genetics to determine whether their tumor is associated with NF1 or is found in isolation (solitary PN); this latter presentation is significantly rarer.
Currently, no preventive treatment is available to prevent the formation of these lesions; however, documenting them early on favors their close surveillance and timely therapeutic decision.
PN is a rare subtype of neurofibroma considered a benign tumor of unknown origin and chronic evolution that presents melanin-producing cells. Clinically, most patients show tumors without visible pigment, but some have hyperpigmentation, hypertrichosis, or both. These lesions appear solitary or associated with NF1. Histologically, their type is diffuse, although some have features of both types (diffuse and plexiform) and are accompanied by melanin-producing cells. As this tumor can be confused with other skin lesions, it is essential to take a biopsy. The histological, ultrastructural, and immunohistochemical findings will allow skin lesion diagnosis and differentiation from other pigmented skin tumors. In case of a new lesion, a multidisciplinary evaluation (Ophthalmology, Neurology, Genetics) should exclude neurofibromatosis. It is necessary to provide genetic counseling to the family upon genodermatosis confirmation. Treatment of PN consists of surveillance and, occasionally, surgical resection.

