











 nueva página del texto (beta)
nueva página del texto (beta) Inglés (pdf)
Inglés (pdf)
 Artículo en XML
Artículo en XML Referencias del artículo
Referencias del artículo
 Enviar artículo por email
Enviar artículo por email Citado por SciELO
Citado por SciELO  Similares en
SciELO
Similares en
SciELO 
 Permalink
PermalinkIntroduction
Peutz-Jeghers syndrome (PJS) is a rare autosomal dominant inherited disease characterized by the development of gastrointestinal polyps, usually hamartomatous type1,2. The incidence of PJS has been estimated at approximately 1 per 50,000 to 200,000 live births2-4. In addition to hamartomatous polyps, patients present with mucocutaneous pigmentation and have been reported to have a nine-fold increased risk of developing gastrointestinal neoplasms than the general population4. Genetic studies have identified, as the main alteration, a mutation in the gene encoding serine-threonine kinase, called STK11, on chromosome 19p13.3. About 70-90% of patients with clinical PJS have a germline mutation in this gene2-9. The clinical manifestations of this syndrome are mucocutaneous pigmentations accompanied by gastrointestinal signs and symptoms, which are secondary to the presence of polyps in the gastrointestinal system10-13.
The diagnosis is confirmed by the following criteria: the presence of two or more Peutz-Jeghers polyps confirmed by histopathology; any number of hamartomatous polyps detected in an individual with a family history of PJS; mucocutaneous pigmentation in an individual who has a family history of PJS; any number of hamartomatous polyps in an individual who also has features of mucocutaneous pigmentations1-3,14.
This study aimed to analyze the epidemiological, clinical, and histopathological data of patients with PJS seen in the Gastroenterology and Nutrition, Surgery, and Anatomic Pathology departments of a tertiary pediatric hospital in Mexico.
Methods
We conducted a retrospective descriptive observational study based on clinical records of patients with an established diagnosis of PJS seen in our institution between 1995 and 2021. A search was conducted for those cases with a PJS diagnosis in the clinical records, identifying them using the International Classification of Diseases, 2010 edition (ICD-10), with the letter Q and numbered 85.8. The data were recorded on a capture sheet: sex, age, family history, the reason for consultation, initial clinical manifestations, nutritional status and complications, and laboratory and histopathological studies results. A descriptive analysis was performed using measures of central tendency.
Results
From 1995 to 2021, we identified 13 records of pediatric patients from unrelated families with a diagnosis of PJS: seven males and six females. Age ranged from 2 to 17 years, with a mean age of 8 years and 10 months at diagnosis, while the median was 7 years and 7 months. A family history of PJS was found in seven cases. The most frequent reason for consultation was abdominal pain in nine patients, where seven were secondary to intussusception. Peculiar pigmentation of the mucous membranes and lips was found in all cases and only in a few cases on the feet, hands, and gums. The rest of the clinical manifestations of these patients are shown in Table 1.
Table 1 Clinical manifestations of Peutz-Jeghers syndrome at diagnosis in pediatric patients
| Findings | n (%) |
|---|---|
| Abdominal pain | 9 (69.23) |
| Intestinal obstruction | 7 (53.8) |
| Rectal bleeding | 7 (53.8) |
| Diagnosis of abnormal nutritional status | 5 (38.4) |
| Constipation | 4 (30.7) |
| Mucous bowel movements | 2 (15.3) |
| Sensation of a mass in the anus | 2 (15.3) |
| Anal prolapse | 1 (7.7) |
At the time of the first endoscopic study, eight patients had polyps in the gastrointestinal tract, mainly of the hamartomatous histological type (Figure 1). In the five patients who did not present polyps at their first evaluation, polyps were demonstrated at subsequent assessment. The most frequent location of polyps detected by colonoscopy was in the descending colon, followed by the rectum (Table 2). All patients underwent resection of technically resectable polyps by endoscopy with a polypectomy snare and electrofulguration; 62 polyps were resected. In 12 patients, the predominant polyps were of the hamartomatous type. Also, polyps of the inflammatory kind, follicular hyperplasia, and adenomatous polyps were found (Table 3 and Figure 1).
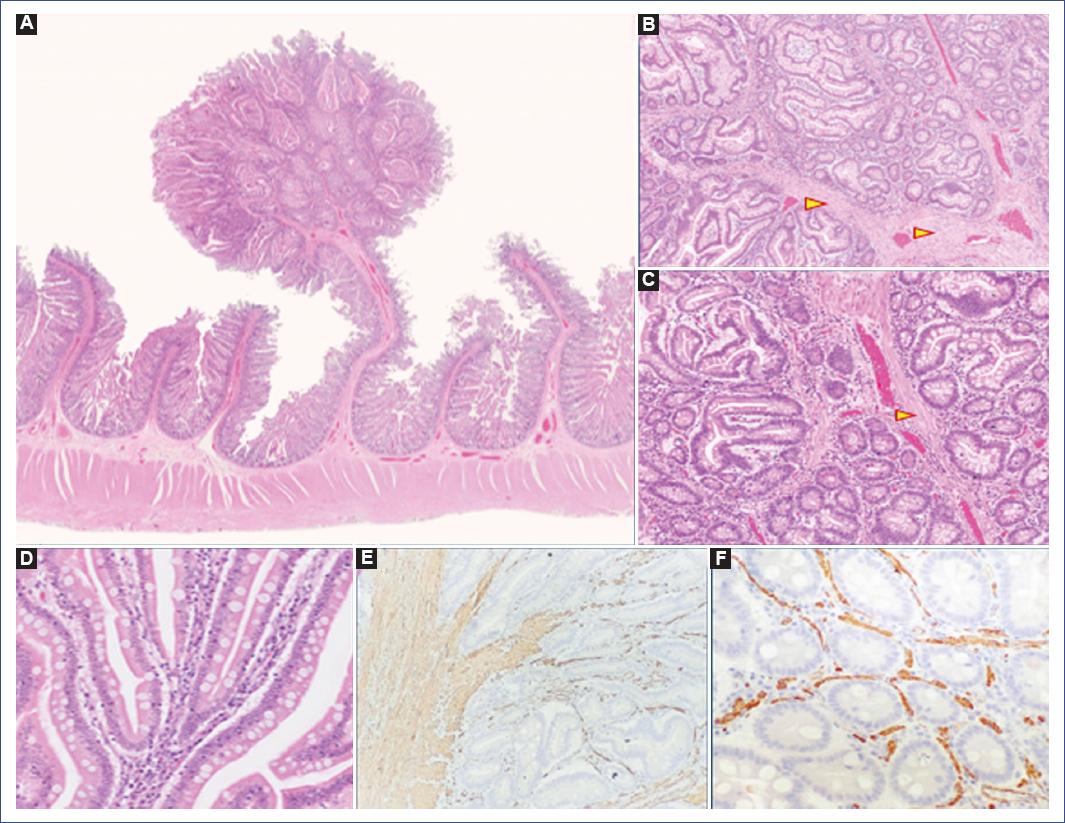
Figure 1 A: pedunculated polyp in the jejunum of a patient with Peutz-Jeghers syndrome in the present series. The polyp is distinguished by being pedunculated with an arborescent pattern. Hematoxylin-eosin stain 1x, showing smooth muscle proliferation and branching (arrowhead). B: hematoxylin-eosin stain 4x. C: hematoxylin-eosin stain 10x. D: the epithelium lining the glands shows no dysplasia. E: immunohistochemistry with smooth muscle anti-actin shows fascicles of smooth muscle in the stroma (10x) and F: between each gland (40x).
From the nutritional perspective, eight of the thirteen patients were eutrophic at the time of consultation, three showed some degree of malnutrition, and two had short stature.
Table 2 Localization of polyps by endoscopy
| Location | Number of polyps | Percentage |
|---|---|---|
| Gastric fundus | 6 | 9.2% |
| Gastric body | 9 | 13.8% |
| Gastric antrum | 5 | 7.6% |
| Duodenal bulb | 2 | 3% |
| Duodenum | 2 | 3 % |
| Jejunum | 7 | 10.7% |
| Terminal ileum | 0 | 0% |
| Ascending colon | 3 | 4.6% |
| Transverse colon | 1 | 1.5% |
| Descending colon | 13 | 20% |
| Sigmoid | 5 | 7.7% |
| Rectum | 12 | 18.4% |
Discussion
Gastrointestinal polyps are an infrequent finding in the pediatric population. However, they are relevant as they may precede or indicate cancer risk. Consequently, a complete evaluation and follow-up with complementary studies and genetic testing are always necessary13,15,16.
PJS is a rare and clinically diverse genetic disease. A 1:1 male-to-female ratio has been described, similar to our study2,17-19. Some authors mention that about 50-70% of PJS patients present a known family history; the rest is considered secondary to new mutations4,20,21. In studies conducted in Uruguay and Korea, family history was reported in 64% and 47% of the participants, respectively20,22. In our series, 53% of patients had a family history of PJS. These patients and their parents should receive genetic counseling.
The main clinical manifestations of this syndrome are mucocutaneous pigmentation associated with abdominal pain and gastrointestinal bleeding secondary to polyps located at any level of the gastrointestinal tract. In most cases, the reason for consultation is a gastrointestinal manifestation, but rarely due to mucocutaneous pigmentation5,11,16,23. Among the clinical manifestations, mucocutaneous pigmentation is found in 66-83% of patients, most frequently in the oral mucosa (94-96%) and often benign. Melanin spots in the mouth may appear on the gums and palate but not on the tongue. Other less frequent areas are the palpebral conjunctiva, fingers, dorsal side of hands and feet, nose, and eyes23,24. These spots usually appear in childhood and reach their maximum expression at puberty. Pigmentation may manifest as scattered or solitary deep brown freckles or spots < 5 mm in diameter21,23,24. In our series, pigmentation was found in 100% of patients, most frequently on the lips and mucous membranes.
The most common cause of consultation is gastrointestinal tract manifestations. The most frequent reason is abdominal pain, although some patients may initially present with symptoms of intussusception6,25. The pain is usually secondary to repetitive episodes of intestinal intussusception, with one of the polyps functioning as the head of the intussusception6,16,17,26. In our series, the most frequent reason for consultation was abdominal pain.
Although this is not the first series of pediatric patients with PJS published in Mexico12,23,27,28, a high prevalence of intussusception was observed in this series(seven cases). Moreover, this was the reason for the initial medical consultation in six patients. This is a relevant finding since almost half of the patients who consulted for the first time did so for acute abdomen due to intestinal occlusion. In one study, intussusception was reported as a severe mechanical complication requiring surgical intervention in 23 of 34 individuals (68%) during pediatric age; 70% of the interventions were emergencies29. These events could be prevented with timely diagnosis, thus avoiding surgical complications (intestinal resection).
Patients with PJS and abdominal pain may present with rectal bleeding and iron deficiency anemia. Rectal bleeding has been reported in 81% of cases2,5; in our study, it was found in 53.8% of participants.
Other infrequent findings are endocrine manifestations: gynecomastia due to increased estrogen levels and the appearance of Sertoli cell tumors30,31. In our study, the finding of these clinical signs was not documented in the clinical records.
The endoscopic phenotypic finding of PJS is the presence of hamartomatous polyps along the gastrointestinal tract, ranging from one to hundreds. Polyps occur in 85 to 100% of cases; most are found in the small intestine (predominantly), colon, and stomach to a lesser extent2,13,14,18. For example, in a series of 182 cases, 96% of the patients had polyps in the small intestine, 60% had colorectal polyps, and 24% had polyps in the stomach32. In other studies, the most frequent location of polyps was in the small intestine as well5,20,23. In our research, the most frequent sites were the descending colon and rectum in seven of the 13 patients studied. Endoscopic evaluation of the small bowel was not performed in all participants because imaging techniques and capsule endoscopy were unavailable to visualize polyps in this segment. Polyps can also be found outside the intestine; for example, in the gallbladder, bronchi, urinary bladder, and ureter1,33,34.
Among the histological findings, 12 of our patients presented hamartomatous polyps; however, the finding of inflammatory polyps, follicular hyperplasia, and adenomatous polyps was notable. Usually, hamartomatous polyps are benign; however, in PJS, they have the potential to develop gastrointestinal and extraintestinal neoplasms35,36. Some authors hypothesize that the malignant potential may be due to a genetic predisposition to mucosal prolapse that underlies the formation of polyps in this syndrome; however, no pathophysiological mechanism has been established to explain the propensity to cancer33.
The American Society for Gastrointestinal Endoscopy and the European Society for Gastrointestinal Endoscopy guidelines recommend prophylactic polypectomy in those patients with polyps > 10 mm observed by enteroscopy. Small bowel polypectomy is considered the first-line treatment in the conservative management of patients with PJS. Polypectomy avoids gastrointestinal bleeding and emergency laparotomy secondary to intussusception; the latter may occur in 16% of patients at 10 years and 50% at 20 years2,12,21,37.
The follow-up of these patients aims to reduce the development of malignant neoplasms and avoid the complications caused by polyps in the gastrointestinal tract, such as obstruction, invagination, and hemorrhage2,6,13,15. Therefore, a complete review of the gastrointestinal tract is advised with the new tools available: double-balloon or video capsule endoscopy, magnetic resonance enterography, and colonoscopy2,5,15,35,38.
The development of malignant neoplasms occurs more frequently after the fourth decade of life. It is estimated that 37 to 93% of patients with PJS will develop cancer at 70. These neoplasms include carcinoma of the colon, small intestine, stomach, and extraintestinal organs, although breast cancer is more frequent (about 50%), and pancreatic, lung, uterus, ovary, and testicular cancer are less frequent8-10,14,39.
Early diagnosis is essential, as affected patients and at-risk family members should receive genetic counseling and close follow-up6,13. PJS patients should be evaluated every year6,15,20,26. Anthropometric and nutritional follow-up of these patients is essential for the early diagnosis of malnutrition; in our study, we identified three patients with some degree of malnutrition and two with short stature.
Follow-up of patients with PJS varies according to symptoms. In asymptomatic patients carrying the mutation, it is suggested to start surveillance by colonoscopy, endoscopy, and small bowel imaging techniques at 8 years. If polyps are found in the initial stage, the evaluation should be repeated every 2 to 3 years. In the case that no polyps are identified in the early studies, surveillance should be repeated at 18 years of age with colonoscopy every 1 to 2 years until reaching 70 years of age and upper gastrointestinal endoscopy at 25 years of age with an interval of 1-2 years, since the risk of cancer begins mainly in adulthood. The adult population should undergo surveillance throughout life due to the risk of developing neoplasms2,6,8,13,15,19,31. It should be considered that nutritional assessment helps to detect possible degrees of malnutrition and allows intervention in the early stages in these patients.
Based on our findings, we conclude the following:
– The main clinical manifestations of PJS in this pediatric population were mucocutaneous pigmentation, abdominal pain, and rectal bleeding.
– PJS should be included as a differential diagnosis in patients with signs and symptoms of bowel occlusion, as intussusception may be initially present in these patients.
– A characteristic finding of PJS is the presence of hamartomatous polyps; however, PJS cannot be excluded if they are not evidenced at the first endoscopy, as some patients initially present with inflammatory polyps, follicular hyperplasia, and adenomatous polyps, exhibiting hamartomatous polyps in subsequent endoscopic studies.

