











 nueva página del texto (beta)
nueva página del texto (beta) Inglés (pdf)
Inglés (pdf)
 Artículo en XML
Artículo en XML Referencias del artículo
Referencias del artículo
 Enviar artículo por email
Enviar artículo por email Citado por SciELO
Citado por SciELO  Similares en
SciELO
Similares en
SciELO 
 Permalink
PermalinkIntroducción
El surfactante es un agente tensioactivo secretado por los neumocitos de tipo II presentes en los alvéolos. Está conformado por un complejo lipoproteico, cuya función fundamental es reducir la tensión superficial de las paredes alveolares, evitando el colapso de los alvéolos al final de la espiración1. Existen trastornos genéticos que alteran el metabolismo del surfactante; aunque son poco frecuentes, pueden causar una morbimortalidad significativa en la población neonatal y pediátrica2,3.
El transportador ABCA3 (ATP binding cassette A3) es una proteína transmembrana encontrada en los cuerpos lamelares de los neumocitos de tipo II, cuya función es el transporte, desde el citosol hacia el interior de los cuerpos lamelares, de las proteínas y los fosfolípidos que conforman el surfactante4,5. Las mutaciones genéticas que afectan la expresión del transportador ABCA3, la síntesis de las proteínas surfactantes B y C, y las mutaciones del gen NKX2-1 originan la enfermedad pulmonar intersticial de gravedad variable en el recién nacido. Debido a la superposición de signos clínicos y radiológicos, es necesario plantearse diagnósticos diferenciales1,4,6.
Aunque se desconoce la verdadera incidencia, las mutaciones del gen que codifica el transportador ABCA3 son la causa más frecuente de disfunción del surfactante7,8. Se ha estimado una frecuencia de presentación de 1/3100 a 1/18,000 en individuos de diferentes grupos étnicos9.
La prueba diagnóstica definitiva para los errores innatos del metabolismo del surfactante es el estudio genético de las mutaciones específicas de estos genes4,10. Sin embargo, las características clínicas y radiológicas de la enfermedad, asociadas con los hallazgos de la biopsia pulmonar en la microscopía óptica, la identificación de las proteínas del surfactante por inmunohistoquímica y el estudio ultraestructural de los cuerpos lamelares de los neumocitos de tipo II mediante microscopía electrónica son de gran ayuda para el diagnóstico de estas disfunciones del surfactante, en especial en países con escasos recursos, donde no se dispone de estudios genéticos para identificar las mutaciones causantes11.
Debido a la infrecuente presentación de los errores innatos del metabolismo del surfactante y el mal pronóstico que, en general, acompaña a esta patología, el objetivo del presente caso fue reportar una adecuada respuesta terapéutica de un recién nacido a término con cuadro clínico, imágenes y estudio de microscopía óptica y electrónica compatibles con una mutación del transportador ABCA3.
Caso clínico
Paciente de sexo masculino, recién nacido a término por parto eutócico con 39 semanas de edad gestacional (primera gestación), con peso al nacer de 2480 g y Apgar 81-95, sin complicaciones durante el parto. A los 3 días de nacido presentó dificultad respiratoria marcada, por lo cual fue intubado y conectado a un equipo de ventilación mecánica; además, se le administró nutrición parenteral durante 2 semanas. Posteriormente dejó la ventilación mecánica y se observó taquipnea persistente, incremento progresivo del esfuerzo respiratorio, hipoxemia persistente y dependencia de oxígeno para mantener una saturación adecuada. Debido a la falta de mejoría clínica, el paciente fue transferido al Instituto Nacional de Salud del Niño de Breña (Lima, Perú) a los 24 días de nacido. No tenía antecedentes familiares de enfermedades pulmonares o congénitas, y tampoco se reportaron intercurrencias durante la gestación.
A su ingreso al servicio de emergencia se le encontró taquipneico, con frecuencia respiratoria de 90 respiraciones por minuto, saturación de oxígeno al 95% y oxígeno suplementario con fracción inspirada de oxígeno al 50%. En la exploración física se le encontró despierto, reactivo a estímulos, con tórax excavado y politirajes. En la auscultación pulmonar se encontraron subcrépitos difusos en ambos hemitórax. El resto de la exploración fue normal.
En los exámenes de laboratorio se encontró en rango normal el título de anticuerpos IgM-IgG para perfil TORCH (toxoplasmosis, rubeola, citomegalovirus, herpes simple), adenovirus y Mycoplasma pneumoniae. La serología para el virus de inmunodeficiencia humana fue negativa. Los hemocultivos seriados fueron negativos. La inmunofluorescencia para virus sincitial respiratorio, adenovirus, influenza tipo A y B y parainfluenza tipo 1, 2 y 3 fue negativa. El dosaje de inmunoglobulinas séricas totales fue normal para IgM, IgG, IgA e IgE. La citometría de flujo con recuento y fenotipo de linfocitos T, linfocitos B y linfocitos NK estaba en rango normal para la edad. También se realizaron fondo de ojo y ecocardiografía, con resultados normales.
La radiografía de tórax y la tomografía espiral multicorte de tórax mostraron compromiso intersticial difuso bilateral con patrón de vidrio esmerilado (Fig. 1).
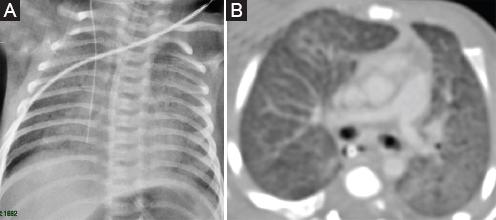
Figura 1 A: Radiografía de tórax en la que se observa un patrón intersticial moderado en ambos campos pulmonares. B: Tomografía de tórax en la que se observa compromiso intersticial difuso bilateral moderado con patrón en vidrio esmerilado y pequeñas imágenes quísticas intraparenquimales (1 mes de edad).
Debido a la hipoxemia y la taquipnea persistente, y ante la sospecha de enfermedad pulmonar intersticial difusa de la infancia, el paciente fue programado para una biopsia de pulmón a cielo abierto. La microscopía óptica de la biopsia de pulmón mostró engrosamiento de los septos alveolares, constituido fundamentalmente por hiperplasia de los neumocitos de tipo II, y la presencia de macrófagos espumosos PAS (ácido peryódico de Schiff) positivos en el interior de la luz alveolar. La microscopía electrónica de la biopsia de pulmón mostró cuerpos lamelares pequeños y anómalos, con presencia de condensaciones electrondensas periféricas, muy características de la deficiencia del transportador ABCA3 (Fig. 2).
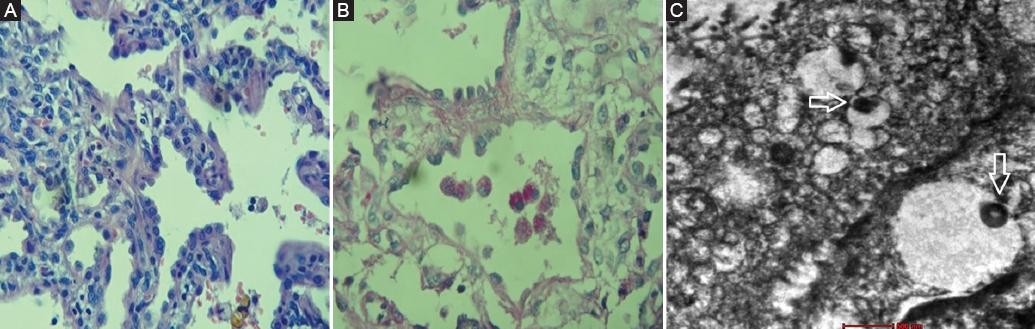
Figura 2 Microscopía óptica. A: Septos alveolares engrosados constituidos fundamentalmente por hiperplasia de neumocitos de tipo II y leve infiltrado inflamatorio linfohistiocitario. Ausencia de fibrosis intersticial (tinción de hematoxilina-eosina, × 200). B: Macrófagos espumosos PAS positivos en el interior de la luz alveolar (× 400). Microscopía electrónica. C: Cuerpos lamelares pequeños y anómalos con presencia de condensaciones electrondensas periféricas concéntricas (flechas) (tamaño de barra: 500 nm).
A los 3 meses de edad, con resultados de la biopsia de pulmón sugerente de disfunción del surfactante, se inició un tratamiento con pulsos de metilprednisolona (dosis de 30 mg/kg al día por 3 días consecutivos), que se repitió mensualmente durante 6 meses y luego fue espaciado con intervalos de 2 meses. Además de los pulsos de metilprednisolona se indicó tratamiento coadyuvante antiinflamatorio e inmunomodulador con hidroxicloroquina (dosis de 10 mg/kg al día fraccionados en dos tomas) y azitromicina (dosis de 10 mg/kg tres veces por semana). Adicionalmente se indicaron corticoides inhalados a dosis altas.
Como la evolución clínica-radiológica fue favorable, el paciente dejó el oxígeno luego del tercer ciclo de pulsos de metilprednisolona. El paciente no requirió atención en la unidad de cuidados intensivos y no presentó infecciones agregadas ni otro tipo de complicaciones, por lo que fue dado de alta. Toleró adecuadamente los pulsos de metilprednisolona, recibiendo seis ciclos mensuales seguidos de seis ciclos bimensuales de manera ambulatoria. Asimismo, toleró la hidroxicloroquina por 18 meses sin complicaciones. Se le realizó un fondo de ojo basal y luego controles periódicos, los cuales fueron normales. Actualmente el paciente se encuentra estable en tratamiento con azitromicina tres veces por semana y corticoides inhalados a dosis altas.
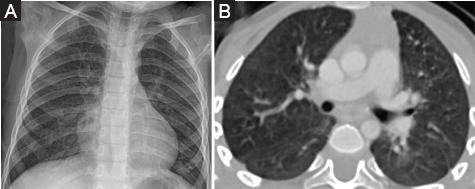
Durante el seguimiento, el paciente ingresó brevemente al hospital por una infección viral intercurrente sin complicaciones, a la edad de 1 año. Actualmente, a los 3 años de edad, se sigue periódicamente en el consultorio ambulatorio de neumología y su evolución clínica y radiológica es estacionaria (Fig. 3).
Discusión
Se presenta el caso de un recién nacido a término, con signos de dificultad respiratoria, cuyas manifestaciones clínicas y radiológicas fueron compatibles con enfermedad pulmonar intersticial difusa.
En los niños menores de 2 años, la causa más frecuente de enfermedad pulmonar intersticial difusa es la disfunción del surfactante o error innato del metabolismo del surfactante. La disfunción del surfactante se origina por mutaciones genéticas que afectan la síntesis de las proteínas precursoras del surfactante o su transporte12,13. La familia de transportadores ABC consta de más de 40 proteínas; la subfamilia ABCA está involucrada en el transporte de fosfolípidos y colesterol, dentro y entre las células. El ABCA3 es un transportador de fosfatidilcolina, el ABCA1 transporta colesterol y fosfolípidos, el ABCA4 transporta fosfatidiletanolamina, el ABCA7 transporta fosfatidilcolina y esfingomielina, pero no colesterol, y el ABCA12 participa en el flujo de lípidos. Por tanto, la familia de transportadores ABCA posee especificidad por determinados sustratos lipídicos. Al tener esta especificidad por sustratos lipídicos, los pacientes con mutaciones en el transportador ABCA3 presentarían una disminuida proporción de fosfatidilcolina en el surfactante pulmonar, además de ciertas características que se pueden identificar por microscopía electrónica, como los cuerpos lamelares anormales, lo que respalda el diagnóstico en el paciente7,11.
El transportador ABCA3 desempeña un papel fundamental en la homeostasis del surfactante pulmonar, pues es el encargado de transportar las proteínas y los fosfolípidos precursores del surfactante desde el citosol del neumocito de tipo II hacia el interior de los cuerpos lamelares y a través de la membrana celular a la superficie alveolar. Las mutaciones del gen ABCA3 son la causa más frecuente de enfermedad pulmonar intersticial difusa secundaria a disfunción del surfactante. La gravedad y el pronóstico de la enfermedad dependen del tipo y la zigocidad de la mutación, de la expresión fenotípica y de factores asociados, por lo que la patología puede manifestarse por primera vez en el niño mayor o el adulto4,5.
Se deben sospechar deficiencias genéticas de las proteínas surfactantes en todo recién nacido a término y en aquellos prematuros que no siguen un curso clínico normal, que presentan dificultad respiratoria progresiva inexplicable, hipoxemia y alteraciones radiológicas difusas, siempre que se hayan excluido causas comunes y de acuerdo con los protocolos internacionales sobre el manejo de la enfermedad pulmonar intersticial en niños. En el paciente de este caso se realizaron diferentes estudios que incluyeron exámenes de laboratorio, radiografía y tomografía de tórax, ecocardiografía y biopsia pulmonar. La biopsia pulmonar proporciona un diagnóstico histopatológico cuando el diagnóstico genético no es concluyente y la progresión de la enfermedad no da tiempo para estudios genéticos, o cuando no se dispone de estos13.
En la tomografía se evidenció compromiso pulmonar intersticial con patrón en vidrio esmerilado y algunos quistes pequeños intraparenquimales, similar a lo reportado por Doan, et al.14 en nueve niños con deficiencia en el transportador ABCA3. Debido a que estos hallazgos no son exclusivos de la deficiencia del transportador ABCA3, fue necesario realizar una biopsia pulmonar para su análisis por microscopía óptica y electrónica; esta última para realizar el estudio ultraestructural de los cuerpos lamelares en el interior de los neumocitos de tipo II15.
Cuando no se dispone de estudios genéticos, las características de las anormalidades de los cuerpos lamelares identificadas mediante microscopía electrónica permiten realizar una aproximación diagnóstica bastante cercana de la mutación del transportador ABCA3 como causa de la disfunción del surfactante11. En los casos de mutación del transportador ABCA3 son característicos los cuerpos lamelares pequeños con membranas de fosfolípidos densamente empaquetados y formaciones electrondensas periféricas (excéntricas), lo que la diferencia de otras disfunciones del surfactante3,10.
Por otro lado, no fue posible realizar el estudio de fosfolípidos y proteínas del surfactante en el líquido del lavado broncoalveolar. Se sabe que los pacientes con mutaciones de ABCA3 presentan una proporción significativamente disminuida de fosfatidilcolina en el lavado broncoalveolar. Si se hubiera realizado este estudio, podría haber orientado el diagnóstico7.
Se conoce que la producción de surfactante aumenta notablemente al final de la gestación y se acelera con los glucocorticoides. Los estudios en modelos animales han demostrado que los glucocorticoides regulan la expresión de la proteína ABCA3 presente en la membrana limitante de los cuerpos lamelares. Este mecanismo se debe a la unión del receptor del glucocorticoide a un sitio específico en la región promotora del gen ABCA3, denominada elemento de respuesta de glucocorticoide (GRE), que induce la activación transcripcional y la subsecuente expresión de la proteína ABCA315. También se han descrito otros factores de transcripción que regulan la expresión del gen ABCA3 en el pulmón, como la proteína de unión a elementos reguladores de esteroles (SREBP) y el factor de transcripción tiroideo-1 (Nkx2.1)16.
La eficacia terapéutica en pacientes con disfunción del transportador ABCA3 es variable. Ocasionalmente se observa una respuesta transitoria a la administración de surfactante exógeno17. No existen ensayos clínicos aleatorizados sobre el tratamiento de la enfermedad pulmonar intersticial difusa en niños; sin embargo, algunas instituciones con experiencia en el manejo de patologías pulmonares en niños recomiendan pulsos de metilprednisolona, prednisona y terapias complementarias con hidroxicloroquina y azitromicina13. Estos medicamentos han sido utilizados en diferentes reportes de casos con una respuesta clínica variable14,17-20.
El paciente del presente caso recibió terapia combinada triple con metilprednisolona, hidroxicloroquina y azitromicina por un periodo prolongado, logrando una mejoría clínica significativa. En la actualidad, el paciente no requiere oxígeno suplementario y se mantiene estable. Sin embargo, el pronóstico a largo plazo es incierto y varía de acuerdo con el tipo de mutación21. En algunos centros europeos se realiza el trasplante combinado de corazón y pulmón en pacientes que no responden al tratamiento2. En Perú no se ha realizado trasplante pulmonar en niños, por lo que debe optarse por el tratamiento médico prolongado.
Los pacientes con esta afección presentan episodios hipóxicos intermitentes, nutrición subóptima y disminución de la estimulación del desarrollo psicomotor, y deben recibir apoyo ventilatorio cuando lo requieran, terapia respiratoria, soporte nutricional y terapia física22.
La mutación del gen que codifica al transportador ABCA3 es una causa infrecuente de enfermedad pulmonar intersticial secundaria a disfunción del surfactante en recién nacidos y niños. La confirmación diagnóstica se consigue mediante estudios genéticos para determinar la presencia de mutaciones en el gen ABCA3. Sin embargo, a falta de estos estudios, la correlación de las características clínicas, radiológicas y tomográficas con los hallazgos en la microscopía óptica y electrónica de la biopsia de pulmón permiten realizar el diagnóstico presuntivo cercano de la mutación del transportador ABCA3.

