











 nueva página del texto (beta)
nueva página del texto (beta) Inglés (pdf)
Inglés (pdf)
 Artículo en XML
Artículo en XML Referencias del artículo
Referencias del artículo
 Enviar artículo por email
Enviar artículo por email Citado por SciELO
Citado por SciELO  Similares en
SciELO
Similares en
SciELO 
 Permalink
PermalinkIntroduction
Osteopetrosis, also known as marble bone disease, is a rare hereditary bone dysplasia characterized by a decreased resorptive activity of osteoclasts leading to inadequate resorption and abnormal bone turnover, causing an increased bone mineral density and increased risk of fractures1. Inadequate bone remodeling produces skeletal deformities, and the lack of bone elasticity coupled with bone fragility increases the risk of fractures2. The endochondral bone in the medullary cavity alters hematopoietic function causing life-threatening anemia, thrombocytopenia, and leukopenia.
Alterations in bone restructuring, which produce a narrowing of the bone canals and limits blood supply, can cause neurological disorders3. Different gene mutations can cause neurological alterations: the mutation of the T-cell immune regulator 1 (TCIRG1) gene causes neuronal compression, while the mutation of the CLCN7 gene causes significant progressive encephalopathy and retinopathy in some patients4.
Clinical suspicion is essential to achieve early diagnosis and timely intervention with allogeneic hematopoietic stem cell transplantation (Allo-HSCT), a curative treatment that can reverse skeletal abnormalities and restore hematopoiesis5.
Epidemiology
Autosomal recessive osteopetrosis incidence is 1 in 250,000 births, with a higher incidence in Costa Rica (3.4 in 100,000 births). In contrast, autosomal dominant osteopetrosis incidence is 1 in 20,000 births6.
Classification
Osteopetrosis has a broad clinical spectrum: from a minimal disease, even asymptomatic, to the severe form known as infantile or malignant osteopetrosis (autosomal recessive), which is life threatening, especially during the first year of life (Table 1).
Table 1 Characteristics of the different types of osteopetrosis and genetic classification
| Population | Inheritance | Bone marrow failure | Diagnosis | Clinical features | Prognosis | Associated genes (% of presentation) | Life expectancy |
|---|---|---|---|---|---|---|---|
| Adult | AD | No | Incidental | Asymptomatic. However, in type I, there is skull affection, and in type II, there is the involvement of the cranial base, vertebrae, pelvis, and long bones. In some cases, bone pain or fractures have been reported | Good | CA II RANKL |
Normal |
| Childhood | AR | Severe | One year of age | Blindness, hearing loss, growth retardation, frequent fractures, severe hypocalcemia, hydrocephalus, neurodegeneration | Poor | TCIRG1 (51-53%)*
CLCN7 (13-16%)** OSTM1 (2-6%)* |
0-10 years 0-3 years 0-2 years |
| Intermediate | AR | Variable | Childhood/ adolescence | Bone pain, arthritis, blindness, hearing loss, osteomyelitis, anemia | Variable | PLEKHMI CLCN7 LRP5 CA II |
Variable |
| Gene and transcript | Location | Variant | Genotype | Classification | |||
| CLCN7 | Exon 5/25 | ENST00000382745.4:c.376C>T
ENSP00000372193.4:p.Arg126Cys Grch37 (hg19) chr16: 1510925 Grch38 (hg38) chr16: 1460924 |
Homozygous | Probably pathogenic variant
Missense variant |
|||
*Percentage of presentation of genes in malignant childhood osteopetrosis.
**Detected in our patient by genetic sequencing.
AR: autosomal recessive; AD: autosomal dominant.
For decades, before identifying gene mutations affecting osteoclasts activity, osteopetrosis had been categorized depending on clinical characteristics and inheritance patterns. The malignant or infantile osteopetrosis with autosomal recessive inheritance pattern is the most severe form and the one with the earliest presentation. Intermediate osteopetrosis also has an autosomal recessive inheritance pattern, and benign or adult osteopetrosis is inherited in an autosomal dominant pattern.
Clinical description
The increase in bone mineral density results in particular physical characteristics, such as macrocephaly, craniofacial deformities, and generalized sclerosis. However, the primary involvement is in the bone marrow and central nervous system. The bone marrows infiltration produces bone marrow failure; thus, hematopoiesis is compromised, and extramedullary hematopoiesis appears, which manifests itself as hepatosplenomegaly.
Etiology
Osteopetrosis is caused by a failure in osteoclasts development and function. Furthermore, mutations in at least 10 genes are found in approximately 70% of patients. This disorder has an autosomal dominant or autosomal recessive inheritance pattern, being the last one the most severe form of presentation7.
Osteoclasts are highly specialized cells derived from mononuclear precursors from the myeloid lineage. Their function is bone resorption, a fundamental process for remodeling and maintaining bone stability and mineral homeostasis.
Some osteopetrosis cases are caused by mutations in genes involved in pH acidification, which depends on the proper function of V-ATPase and CLCN-7. Homozygous mutations in these genes can cause malignant osteopetrosis.
Diagnosis
The diagnosis of childhood osteopetrosis is based on clinical and radiographic evaluations and confirmed by genetic testing2. It is essential to consider the diagnosis based on clinical suspicion and corroborate it by radiography and image studies, where cortical atrophy and generalized sclerosis mainly affecting the skull, pelvis, and spine can be appreciated. The characteristic image of bone within a bone, particularly in vertebrae and phalanges, can be observed. Age of presentation, inheritance, and associated clinical features such as pancytopenia, neurodegeneration, mental retardation, tubular acidosis, neuronal compression, and visual and auditory impairment support the diagnosis.
In case of suspicion, laboratory studies should be requested, such as serum calcium, parathyroid hormone, phosphorus, creatinine, 25-hydroxyvitamin D, and lactic dehydrogenase levels, which provide mineral homeostasis data. Also, a complete blood count with differential is necessary for bone marrow failure evaluation. Bone biopsy is useful for differentiating rich or deficit osteoclasts forms of osteopetrosis; however, it should not be performed routinely and is not essential for diagnosis.
Creatine kinase BB isoenzyme has been detected in patients with malignant osteopetrosis with CLCN-7 mutation; however, its levels do not correlate with the severity of the disease, and normal levels do not exclude the diagnosis8.
Finally, the specific mutation must be confirmed by genetic testing to provide follow-up, prognosis, a probable response to treatment, recurrence, and genetic counseling.
Multidisciplinary management of these patients is vital since multiple organs and systems are affected. Endocrinologically, serum calcium levels should be monitored since bone cannot mobilize calcium due to the osteoclasts defect, which causes hypocalcemia and, secondary to this, tetany and seizures.
It is necessary to periodically evaluate patients visual capacity to detect and control the emergence of major visual complications, such as optic nerve atrophy. Patients often have dental abnormalities, such as delayed or failed tooth eruption, malformation, and easy teeth loss. Fractures and their complications with osteomyelitis are frequent. Therefore, an expert orthopedic surgeon should evaluate patients. Neurologically, developmental delay, compression neuropathies, and craniofacial deformities should be intentionally sought.
Regarding hearing, nerve compression can lead to sensorineural deafness. Concerning hematological complications, pancytopenia is the primary life-threatening condition of patients with osteopetrosis, leading to severe infections and bleeding5.
Differential diagnosis
The differential diagnosis should be made with any disease that causes bone sclerosis, such as berylliosis, myelofibrosis, Pagets disease, and malignant diseases. X-rays may show an increase in bone mineral density; however, compared to osteopetrosis, this alteration improves overtime. When osteopetrosis is diagnosed, it is essential to distinguish the subtype because each has a different evolution, progression, prognosis, and treatment.
Genetic alterations
The primary characteristic mutation of osteopetrosis is found in the TCIRG1 gene, which encodes the a3 subunit of the vacuolar H+ATPase (V-ATPase), which pumps protons through the membranes, helping to regulate cellular and extracellular pH.
The V-ATPase plays an essential role on the surface of osteoclasts. This protein is embedded in a membrane that contacts the bones surface and forms a compartment between the osteoclast and the bone surface. Its function is to pump protons into the bone, acidifying it and creating a suitable bone resorption environment9. This mutation is responsible for 50% of patients diagnosed with recessive osteopetrosis10.
The CLCN7 gene encodes a chloride channel (CLC-7) that provides electroneutrality during the acidification process4. The mutation of this gene is present in 10-15% of patients with malignant osteopetrosis.
The third gene involved in this disease is OSTM1, which causes a more aggressive disease phenotype, shortening survival.
Treatment
HSCT is a procedure in which stem cells (HSC) from a disease-free donor are infused into patients with the disease. This procedure is performed to restore bone marrow function. There are different types of HSCT, depending on the type of HSC donor: autologous transplant (donor and recipient are the same subject), allogeneic transplant, which is performed from a donor with human leukocyte antigen (HLA) 100% compatible with the recipient and can be a related donor (family member) or an unrelated donor (non-family member), and haploidentical transplant, in which the family donor has an HLA 50% compatible with the recipient.
The initial treatment of osteopetrosis is mainly supportive and focuses on treating the main complications, such as pancytopenia, infections, and bleeding.
Fractures and arthritis require specialized treatment and follow-up since there is usually a delay in consolidation, osteomyelitis, and recurrent fractures.
Developmental delay and seizures associated with normal calcium levels often indicate childhood osteopetrosis. Neurological evaluations, including magnetic resonance imaging (MRI) and electroencephalogram, periodic ophthalmological examinations, including visual evoked potentials, and fundus examination to detect optic nerve atrophy, should be performed as soon as possible.
Hearing function evaluations should also be performed because, initially, neurosensory damage is mild; however, it may contraindicate HSCT as it progresses to severe forms.
Bone marrow failure can be treated initially with platelet and red blood cell concentrates transfusions; however, the definitive treatment of osteopetrosis is Allo-HSCT. In this pathology, HSCT is considered a therapeutic emergency because, as the disease progresses, sensory, visual, and hearing alterations become irreversible if treatment is late.
Transplant-related complications include transplant rejection, delayed graft function, hepatic sinusoidal occlusive syndrome, pulmonary hypertension, and hypercalcemic crises11.
It is necessary to keep this disease under consideration when evaluating a patient with multiple dysmorphia, retarded growth and development, as well as sensory alterations, accompanied by metabolic and hematological alterations since timely diagnosis will allow to perform an effective treatment and thus avoid a progression of the disease with more significant neurosensory impairment. Highlighting the importance of this pathologys treatment, we present the case of a patient with osteopetrosis treated with an Allo-HSCT, who had a successful evolution, even with a late disease diagnosis and a mild neurosensorial impairment.
Clinical case
We present the case of a 3-year and 2-month-old male patient from the State of Mexico. In his family medical history, healthy parents with no chronic degenerative diseases or consanguinity, a healthy 8-year-old sister, and no history of cancer, genetic or metabolic diseases, or immunodeficiency were reported.
At 1 year of age, the patient attended the Hospital Infantil de México Federico Gómez (HIMFG) for presenting community-acquired pneumonia (CAP), and later required hospitalizations for the same diagnosis (Fig. 1).
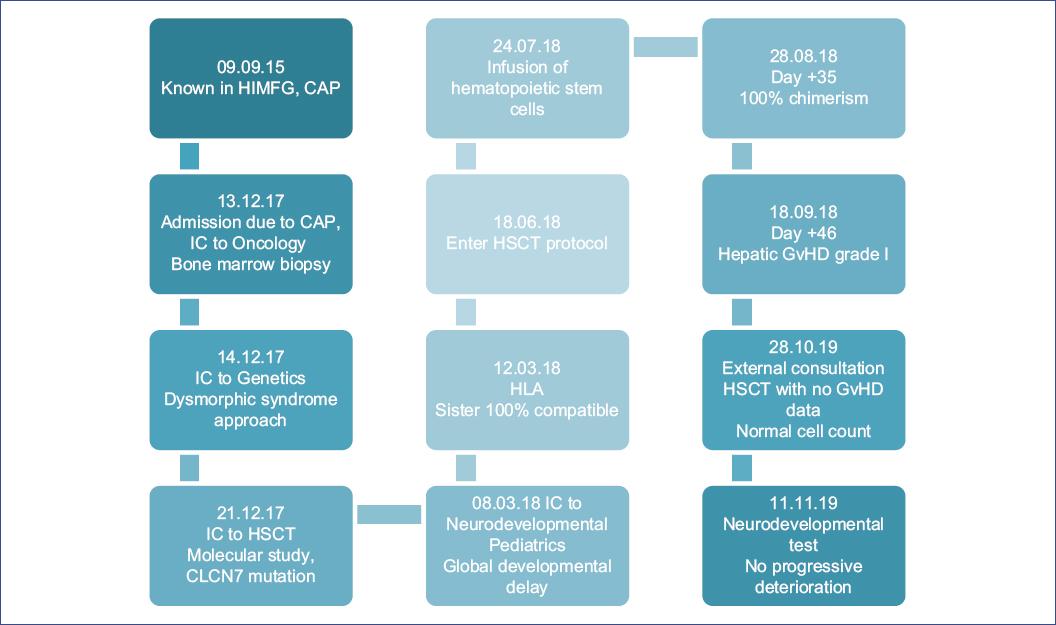
Figure 1 Chronological evolution of a patient with osteopetrosis. CAP: community-acquired pneumonia; GvHD: graft-versus-host disease; HIMFG: Hospital Infantil de México Federico Gómez; HLA: human leukocyte antigen; HSCT: hematopoietic stem cell transplantation; IC: interconsultation.
At 3 years of age, he was admitted again for CAP. On physical examination, generalized integument paleness, macrocephaly, hypertelorism, microretrognathia, prominent forehead, telecanthus, trichiasis, prominent eyes, flat nasal bridge, short neck, and narrow chest were observed. The skull X-ray showed the mask sign, characterized by generalized bone hyperdensity, predominantly at the base of the skull, which gave it the appearance of a mask in the orbital region. A chest X-ray was performed, observing an increase in bone density at the rib, sternal, and vertebral level. In addition, the bone within a bone sign, which consists of lines parallel to the tibias cortical bone, was observed in the long bone X-ray. In laboratory studies, bicytopenia was reported in the complete blood count (hemoglobin 5.7 mg/dl, hematocrit 17.7%, leukocytes 13,400 cells/μL, total neutrophils 4020 cells/μL, and platelets 22,000 cells/μL). Therefore, as part of the approach, a bone marrow biopsy was requested, which reported hypocellularity, small intertrabecular spaces with fibrous tissue, and endochondral ossification. Due to the presence of dysmorphic syndrome, the patient required evaluation by the genetics service.
The patient was diagnosed with osteopetrosis at 3 years and 2 months of age, and as part of the evaluation by the HSCT service, the molecular study was carried out using second-generation sequencing in the HIMFG Research Unit. A mutation in the CLCN7 gene was reported, and the patient was classified as a homozygous genotype (Table 1). With this mutation, the patient met the criteria for an Allo-HSCT.
During hospitalization, the patient presented bilateral serous otitis media with secondary conductive hearing loss, for which ventilation tubes were placed. Influenza was detected, requiring antiviral treatment; another event of otitis media with otorrhea occurred, for which the patient required myringotomy and placement of ventilation tubes again.
A skull MRI was performed in the pre-transplant evaluations, where frank cortical atrophy with secondary ventricular dilation associated with its underlying pathology was observed. We detected a slight alteration in the bilateral visual pathways conduction, together with a normal audiometry report, employing visual evoked potentials. Due to these results and the significant risk of developmental delay, the Infant Development Assessment test was performed, in which the patient met the criteria for risk of developmental delay. In the Battelle developmental test, a total development ratio of 63 was reported, corresponding to an equivalent age of 2 years and 3 months of age (deficit of 1 year and 5 months [37%]). Finally, the patient was diagnosed with global neurodevelopmental delay.
According to the HSCT protocol of the HIMFG, comorbidities were evaluated by the following services: ophthalmology, endocrinology, cardiology, infectology, otorhinolaryngology, pulmonology, nephrology and gastroenterology, and nutrition. Neurosensory, severe visual, or hearing damage were ruled out. In the admission blood count, before the HSCT, the following results were reported: total leukocytes 5300 cells/μL, total neutrophils 2070 cells/μL, platelets 35,000 cells/μL, and hemoglobin 9.1 mg/dL. At 3 years and 9 months of age, Allo-HSCT was performed from 100% compatible related donor (sister), using a myeloablative conditioning regimen: on days −7, −6, −5, and −4, the patient received busulfan (1.2 mg/kg/day), and on days −3 and −2, cyclophosphamide (60 mg/kg/day). Subsequently, a dose of 4.7 × 107 cells/kg obtained from bone marrow was infused.
As part of the prophylaxis for graft-versus-host disease (GvHD), immunosuppression was used based on cyclosporine (CsA, 3 mg/kg/day) and methotrexate (15 mg/m2 day +1 and 10 mg/m2 day on days +1, + 3, +5, +7, and +11). However, due to the lack of graft data on day +22, the calcineurin inhibitor dose was decreased, continuing with stimulation using granulocyte stimulating factor (Filgrastim). On day +27, granulocytic graft data with a total neutrophil count of 1200/μL were observed. On day +35, the patient showed 100% chimerism. Therefore, the dose of Filgrastim was decreased, and treatment with platelet-stimulating factor (Eltrombopag) was added, which continued for 6 months.
During this period, the patient developed pneumonitis requiring supplemental oxygen support through nasal prongs and inhaled steroid managementand presented hypercalcemia and hyperphosphatemiawhich responded to management with hydration, forced diuresis, and phosphorus chelator, with no other complications. Subsequently, on day +46, Grade II acute GvHD was diagnosed, supported by altered liver function tests with elevated bilirubin, for which treatment with methylprednisolone (1 mg/kg/day), cyclosporine, and ursodeoxycholic acid was administered. The treatment was gradually decreased according to the clinical course and CsA levels. Due to the adequate response to treatment, it was possible to withdraw immunosuppressive drugs (currently, the patient is not immunosuppressed).
According to the follow-up protocol, a periodic quantification of chimerism was carried out (on days +15, +21, +28, +35, +100, and +150) to verify the transplant graft. On day +15, a chimera of 16.6% was found; subsequently, on day +21, the percentage increased to 59.88%, and on days +35, +100, and +150 after HSCT, 100% chimerism was documented (Fig. 2). At present, at 5 years of age, the patient continues in monthly outpatient follow-up and is in the process of entering preschool education. Furthermore, cell numbers are normal, with no platelet-stimulating factor or erythropoietin requirement, no GvHD data, and deferasirox treatment to avoid iron overload. Due to delayed growth, a weight of 14.2 kg (PZ −2), and a height of 90 cm (PZ −3) at 5 years and 3 months of age, the patient is being followed up by the endocrinology service. The patient maintains a visual capacity with a slight deficit but no progression and under ophthalmology service follow-up.

Figure 2 Laboratory studies of the patient with post-transplant osteopetrosis. A. Hemoglobin value after transplantation. B. Molecular chimerism after transplantation. C. Platelet value after transplantation. D. Granulocytic value after transplantation.
In addition, the patient undergoes an annual audiological evaluation, with no apparent deficit. As part of the follow-up, the EDI test that reported significant developmental delay improved compared to the previous results. An increase in the score of six evaluated domains was observed, finding that the personal-social and adaptive sphere significantly improved. Since the patient showed no added neurological deficit and an improvement was reported in neurodevelopmental tests, a new neurological imaging study was not considered necessary at the time. At present, the patient is being monitored by the neurodevelopment and neurology service.
Discussion
Infantile osteopetrosis is a rare hereditary disease, with a characteristic phenotype of short stature, hypocalcemia, fractures, compression neuropathy, and pancytopenia; it is caused by the failure in the development and function of osteoclasts. We present the case of a 5-year-old male with a CLCN7 gene mutation and a 1-year follow-up after Allo-HSCT. Because osteopetrosis is caused by a defect in the osteoclasts derived from the hematopoietic cell line, bone marrow transplantation is the definitive treatment. Our patient underwent Allo-HSCT at 3 years and 9 months of age and currently has normal cell counts. The skeletal deformities and bone sclerosis shown on the X-rays improved since treatment (Fig. 3). Since the first approach, this patient exceeded the 3-year life expectancy reported for osteopetrosis patients with the CLCN-7 mutation (Table 1). Over 600 days after the transplant, the patient has shown a favorable evolution, decreasing the need for transfusions and hospitalization. Furthermore, skeletal deformities and neurodevelopmental tests have improved, leading to a better quality of life.
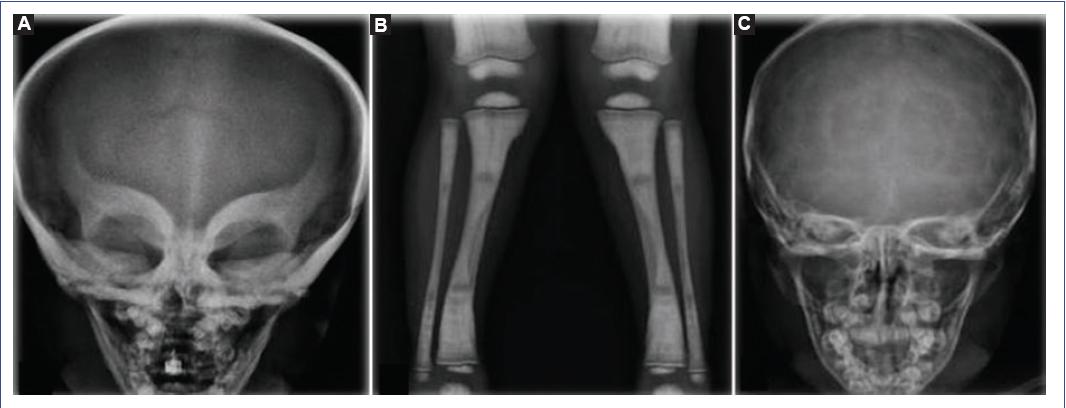
Figure 3 Osteopetrosis patient imaging studies. A. X-ray of the skull showing mask sign. Predominant generalized bone hyperdensity is observed at the base of the skull (before transplantation). B. X-ray of tibia and fibula with the bone within a bone sign. C. X-ray of the skull after transplantation with decreased bone hyperdensity.
Severe osteopetrosis with increased bone mineral density results from the absence of osteoclasts, caused by a defect in the differentiation of osteoclast precursors or by dysfunctional osteoclasts. Therefore, the definitive treatment must be the transplant of hematopoietic stem cells with a bone marrow origin to obtain bone marrow-derived osteoclasts.
Abnormal bone expansion interferes with hematopoiesis, causing life-threatening pancytopenia and extramedullary hematopoiesis in the spleen and liver, and frequent hepatosplenomegaly. This patient presented pancytopenia at diagnosis. Initially, an infiltrative syndrome was suspected but ruled out; later, osteopetrosis was diagnosed.
Patients with some mutation usually have neurosensory, visual, and auditory alterations caused by bone compression12. In this patient, cortical atrophy accompanied with secondary ventricular dilation and bilateral visual pathway conduction alteration was documented, consistent with some reports in which MRIs show myelination delay and progressive cortical and subcortical atrophy13. The leading causes of death in these patients are infections, bleeding, and severe anemia, and allogeneic hematopoietic progenitor cells transplant is the only curative option14. A 5-year survival rate of 73% is reported when 100% compatible donor transplant is performed15. Risk of death increases in the first 10 years if Allo-HSCT is not performed due to the suppression of hematopoiesis in the bone marrow and its consequences.
The highest survival is reported in patients who received a transplant 100% HLA compatible of a related donor. The 5- and 10-year survival in patients with 100% compatible related donor Allo-HSCT is 62% in both, while for transplant of unrelated donor decreases to 42% and 39%, respectively. Therefore, it is of critical importance to obtain a related donor Allo-HSCT16. As for the complications that can occur after transplant in patients with osteopetrosis, primary and late graft failure has been described. In the present case, we observed delayed platelet engraftment with an adequate response to the platelet-stimulating factor. This complication imposes a higher mortality risk in transplanted patients17.
The leading cause of transplant-related mortality is graft failure, accounting for 50% of deaths in transplants from related donors and 43% from unrelated donors.
Allo-HSCT improves bone lesions within 2 months, and they resolve entirely within a year of transplant18. However, survivors reported that only 7% experience improvement regarding the optical damage, 69% show no progression, and 25% show deterioration.
This clinical case generates essential information about HSCT and osteopetrosis despite the age at the time of diagnosis because the evolution has been favorable. Therefore, we emphasize the importance of clinical suspicion for timely diagnosis and treatment. As no documented evidence exists in our country, it is imperative to generate this kind of reports due to the remarkable improvement that a timely treatment means in patients quality of life and survival.
There is only one previous report in our country, documenting a 9-month-old patient who required a second transplant at 13 months of age, using the umbilical cord as a cellular source on both occasions19. It is necessary to continue investigating and reporting these cases to enrich this knowledge.
In this case, we emphasize the importance of early diagnosis. Suspicion of the disease in a patient with pancytopenia, craniofacial and thoracic dysmorphia, hepatosplenomegaly, and radiological evidence of bone sclerosis should prompt a comprehensive approach and early osteopetrosis diagnosis. Therefore, timely interventions and early treatment with Allo-HSCT could be established to improve the patients clinical conditions. It is essential to perform an Allo-HSCT, preferably before 1 year of age, to avoid progressive and irreversible neurological damage and decrease mortality. It has been shown that patients who have 100% compatible donor for Allo-HSCT have a 5-year survival of 73%. However, with an unrelated donor, survival of 40% has been reported.
Timely Allo-HSCT has an impact on the patients prognosis and quality of life. It is possible to perform late Allo-HSCT in a patient with mild neurosensory deficits and achieve a cure for osteopetrosis, stop the progression of neurological damage, and achieve an adequate quality of life on the condition where a multidisciplinary treatment is available.

