











 nueva página del texto (beta)
nueva página del texto (beta) Español (pdf)
Español (pdf)
 Artículo en XML
Artículo en XML Referencias del artículo
Referencias del artículo
 Enviar artículo por email
Enviar artículo por email Citado por SciELO
Citado por SciELO  Similares en
SciELO
Similares en
SciELO 
 Permalink
PermalinkIntroducción
Los linfomas no Hodgkin (LNH) son un grupo de neoplasias heterogéneas derivadas de las células linfohematopoyéticas. Rara vez se presentan antes de los 2 años, ya que la edad de presentación más común es entre los 5 y los 15 años1,2. La prevalencia es mayor en el sexo masculino y la incidencia es mayor en los hispanos2. La principal característica de estas neoplasias es la proliferación anormal y descontrolada de células precursoras que presentan translocaciones en cromosomas, lo que contribuye a una producción anormal de proteínas de fusión que afectan los mecanismos de control del crecimiento y la maduración de dichas células1. Gracias a las técnicas de biología molecular, la Organización Mundial de la Salud las ha clasificado en diferentes subgrupos (Tabla 1).
Tabla 1 Clasificación de la Organización Mundial de la Salud de los linfomas no Hodgkin (2008)2
| Subtipo de linfoma | Frecuencia |
|---|---|
| Neoplasias de precursores linfoides | |
| Linfoma linfoblástico T | 15-20% |
| Linfoma linfoblástico B | 3% |
| Neoplasias de células B maduras | |
| Linfoma de Burkitt | 35-40% |
| Linfoma difuso de células grandes B | 15-20% |
| Linfoma de células B mediastínico primario | 1-2% |
| Neoplasias de células T maduras | |
| Linfoma anaplásico de células grandes, cinasa del linfoma anaplásico (ALK) positivo | 15-20% |
Dentro de los LNH, el linfoma linfoblástico (LL) suele presentarse como una enfermedad supradiafragmática, que puede invadir fuera de la cavidad torácica3. Dependiendo de la estirpe precursora, la sintomatología varía. En el caso de las células T (15-20% de los linfomas), se presentan con una masa adenopática de rápido crecimiento en la región cervical o mediastínica, usualmente con compromiso de la vía aérea o de la vena cava. Por otro lado, existe afectación de los ganglios periféricos, los huesos y la piel, y en caso de LL, de precursores B (3% de los linfomas), como en el presente caso1.
El objetivo de este reporte es divulgar información acerca de una enfermedad poco descrita en la literatura y con una expresión poco habitual. Es necesario que tanto especialistas como médicos del primer nivel de atención conozcan estos datos para que puedan tomar las decisiones adecuadas cuando se encuentren con un caso similar.
Caso clínico
Paciente de sexo masculino, de 1 año y 11 meses, originario del Estado de México, sin antecedentes de importancia. Inició su padecimiento presentando marcha claudicante a expensas del miembro inferior derecho, sin sintomatología agregada (dolor, irritabilidad, artralgias ni fiebre). Dos semanas más tarde, la marcha claudicante se tornó bilateral, sin presentar ninguna otra sintomatología asociada con el cuadro. Fue valorado por un ortopedista particular, quien diagnosticó pes planus y dio tratamiento con zapatos ortopédicos. Dos meses después, el paciente refirió dolor en los miembros inferiores que impedía la marcha y se agregaron síntomas de hipotonía generalizada, aumento de volumen de las muñecas con flogosis y deformidades induradas en ambas muñecas. Posteriormente se agregaron al cuadro clínico artralgias localizadas en los tobillos con irradiación hasta el tercio inferior de las piernas, con eritema, aumento de volumen y deformidad proximal de ambas tibias. La madre negó sintomatología como fiebre u otras manifestaciones. Más tarde, el paciente presentó limitación a la movilidad del brazo derecho y, al no presentar mejoría, acudió a un ortopedista pediatra, quien solicitó radiografías de huesos largos. En estas radiografías se observaron lesiones osteolíticas sin rotura de la cortical, principalmente metafisaria distal de fémur y tibia (Figura 1).

El paciente acudió a una institución de tercer nivel para evaluar la posible presencia de un tumor en las extremidades o algún proceso infeccioso. La valoración por parte del servicio de reumatología mencionó que se trataba de una probable artritis juvenil, por lo que el paciente recibió tratamiento con antiinflamatorios no esteroideos y se indicó continuar en control de consulta externa. Sin embargo, se perdió el seguimiento.
Dos meses después, el paciente acudió a una institución de tercer nivel diferente, presentando conjuntivitis, astenia y adinamia subjetiva, e incapacidad para movilizarse por dolor en los miembros inferiores que impedía la marcha.
En la exploración física, los datos fueron los siguientes: peso 9.65 kg (percentil 2; Z −1.99), talla, 80.1 cm, índice de masa corporal 15, brazada 89 cm, longitud del pene 7.5 cm, circunferencia del pene 5.5 cm, índice volumen del pene 18.06 (Z +5.73), volumen testicular derecho 9.5 cm3 (+13.45) y volumen testicular izquierdo 9.5 cm3 (+13.2).
Presentó palidez generalizada, afebril, respiración oral persistente. Aplanamiento del puente nasal con filtro alargado, orofaringe con hipertrofia de amígdalas de grado ll-lll, múltiples adenopatías en la región cervical anterior y posterior bilaterales, móviles, no dolorosas, sin eritema. En la región axilar se identificaron campos pulmonares sin alteraciones. Abdomen blando y depresible. Se palpó hepatomegalia de 3 cm por debajo de reborde costal derecho y esplenomegalia de 2 cm por debajo del borde costal izquierdo. Se encontraron lesiones cutáneas caracterizadas por nódulos móviles no adheridos de 1 cm en la cara anterior del brazo y en la región escrotal, del mismo color que el resto de la piel. Miembros pélvicos con deformidades a expensas de tejidos blandos. Extremidades superiores e inferiores con evidente malformación en los tercios distales. El paciente refirió dolor 8/10 cuando se le solicitó que se moviera o cambiara de posición.
Ante la sospecha de activación de la pubertad, el paciente fue referido al servicio de endocrinología, donde se le realizaron estudios de gonadotropinas, testosterona, biometría hemática, química sanguínea, electrolitos séricos y perfil hepático, obteniendo los resultados siguientes: alfa-fetoproteína, 2.5 Ul/ml; gonadotropina coriónica humana-beta libre, < 2 ng/dl; hormona luteinizante (LH) basal, 0.221 mU/ml; LH a los 180 minutos del estímulo con leuprorelina acuosa (análogo de la hormona liberadora de gonadotropina), 4.3 mU/ml, LH a las 24 horas del estímulo, 1.44 mU/ml; testosterona basal, < 20 ng/dl; testosterona a los 180 minutos del estímulo con leuprorelina acuosa, < 20 ng/dl, y testosterona a las 24 horas del estímulo, 20 ng/dl. La edad ósea se encontró en concordancia con la edad cronológica del paciente (2 años). Con estos resultados, se descartó el diagnóstico de pubertad precoz y se consideró macrogenitosomía.
Los resultados de la química sanguínea fueron los siguientes: ácido úrico, 6.7 mg/dl; nitrógeno ureico en sangre, 20.2 mg/dl; creatinina, 0.39 mg/dl; glucosa, 105 mg/dl, y urea, 43.2 mg/dl. La biometría hemática mostró valores de hemoglobina de 11.8 g/dl, 34.9% de hematocrito, 13.3 × 103/mm3 de leucocitos, 41% de neutrófilos, 48.4% de linfocitos, 9.2% de monocitos, 0.9% de eosinófilos y 426 × 103/mm3 de plaquetas. Los resultados de los electrolitos fueron los siguientes: Na, 137 mEq/l; K, 4.4 mEq/l; Cl, 105 mEq/l; P, 6 mEq/l; Mg, 2.2 mEq/l, y Ca, 11.7 mEq/l.
También se cuantificó el calcitriol (1,25-vitamina D) y se reportó un valor de 31.1 pg/dl. Finalmente, los resultados del perfil hepático fueron los siguientes: fosfatasa alcalina, 144 UI/l; deshidrogenasa láctica, 490 UI/l; aspartato aminotransferasa, 116 UI/l; alanina aminotransferasa, 78 UI/l; gamma-glutamil transpeptidasa, 12 UI/l, y creatina cinasa, 24 UI/l.
De acuerdo con esta evaluación, se detectó hipercalcemia y se solicitó valoración con imagenología. En el abordaje radiológico se reportaron zonas líticas de huesos largos de aproximadamente 2 meses de evolución (Figura 1), sin afección de columna ni de cráneo. Dada la presentación clínica y paraclínica, se presumió el diagnóstico de displasia ósea y probable síndrome de McCune-Albright, que se caracteriza por afectación cutánea, displasia fibrosa del hueso y pubertad precoz.
Además de las radiografías, se realizó una tomografía por emisión de positrones (PET) (Figura 2), la cual reportó zonas de destrucción e hipercaptación con predominio en el área maxilar, sugiriendo un proceso maligno y desplazando el diagnóstico de displasia ósea. La PET mostró una infiltración más clara en el esqueleto apendicular y el macizo facial y mandibular, con lesiones de aspecto infiltrativo, condicionando disrupción de la cortical ósea. El nivel del bazo (eje longitudinal de 6.9 cm y valor de PET semicuantitativo [SUVmax] de 3.0 cm) se encontró en correlación con el hígado (SUVmax de 1.2 cm). En cuanto a las lesiones esqueléticas, la más metabólica fue de 3.2 cm en la cabeza humeral izquierda. El hígado se encontró disminuido en densidad y vascularidad, y con aumento del metabolismo y de su tamaño. Se encontraron, además, ganglios cervicales de 10.4 mm en su eje corto.

Figura 2 Se evidencian lesiones hipermetabólicas en el esqueleto apendicular y el macizo facial, sugestivas de infiltración ósea de linfoma primario conocido (linfoma linfoblástico), con importante afección esplénica.
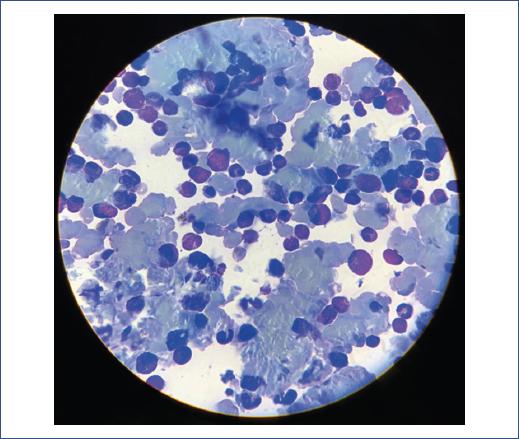
Con base en los hallazgos clínicos y paraclínicos, se consideró histiocitosis de células de Langerhans sistémica, ya que esta suele afectar a niños menores de 3 años con manifestaciones clínicas como astenia, adinamia, adenopatías y hepatoesplenomegalia por infiltración. En la serie ósea se encontraron múltiples lesiones osteolíticas bien definidas y reacción perióstica en los huesos del miembro pélvico izquierdo. Continuando con la evaluación del paciente, el servicio de dermatología describió dermatosis en la región supraciliar derecha y la región preauricular, con dos neoformaciones subcutáneas irregulares, mal definidas, discretamente eritematosas, de consistencia formes y móviles, de 1.5 cm, de 1 mes de evolución. A partir de la biopsia de un ganglio inguinal, el servicio de histopatología reportó linfoma/leucemia de precursores B, por lo que se realizó aspirado de médula ósea, cuyos resultados fueron los siguientes: blastos, 9%; eosinófilos, 7%; linfocitos, 9%; promielocitos, 5%; mielocitos, 5%, y bandas, 1%. El líquido cefalorraquídeo fue negativo a la presencia de blastos en análisis por citocentrífuga. Se descartó la infiltración al sistema nervioso central. La biopsia de médula ósea reportó infiltración con desoxinucleotidil-transferasa terminal positiva, factor de transcripción exclusivo de células B (PAX5) positivo, mieloperoxidasa negativa, con expresión de marcadores para precursores B sin marcadores para precursores mieloides. Se concluyó el diagnóstico de LL preB en estadio IV (Figura 3). Se decidió, entonces, iniciar el tratamiento para LL de células B de acuerdo con el protocolo BFM 90.
Discusión
El presente caso tiene elementos que lo diferencian de una presentación común de LL de precursores B. En primera instancia, la edad de presentación del paciente es infrecuente para los linfomas, ya que suele ser entre los 5 y 15 años1, y la estirpe celular también es poco común. En segundo lugar, el estímulo iatrotrópico fue el dolor óseo. Los demás datos fueron descubiertos incidentalmente. Asimismo, se debe considerar el hecho de que la evolución clínica del padecimiento fue de 3 meses, a diferencia de los LNH convencionales, que suelen ser de progresión rápida e insidiosa, con tendencia a manifestarse en forma de urgencia oncológica. Otro dato de importancia es la presencia de adenopatías generalizadas, que en un inicio desviaron la consideración diagnóstica de un LNH, por lo que se estimaron otros posibles diagnósticos.
Durante el abordaje del paciente se detectó hipercalcemia, alteración metabólica que, en presencia de cáncer, se considera un síndrome paraneoplásico. Este fenómeno en niños es excepcional4. A diferencia del síndrome de lisis tumoral, en el que es característica la hipocalcemia secundaria a la hiperfosfatemia, se ha postulado que la hipercalcemia como síndrome paraneoplásico es secundaria a actividad neoplásica, y representa una urgencia oncológica debido a que pone en riesgo la vida del paciente. Ocasiona síntomas variados, desde náuseas, falta de concentración, poliuria-polidipsia y astenia, hasta en casos graves disfunción del sistema nervioso central, alteraciones cardiovasculares (hipertensión arterial, acortamiento del intervalo QT en el electrocardiograma, taquiarritmias e incluso paro cardiaco) e insuficiencia renal5. La hipercalcemia es sumamente rara. Se presenta en el 0.4-1.3% de los cánceres en la edad pediátrica y se ha reportado en rabdomiosarcoma, hepatoblastoma, linfoma de Hodgkin y no Hodgkin, tumores cerebrales, neuroblastoma, angiosarcoma, leucemia linfoblástica aguda y leucemia mieloide6. No se ha descrito hipercalcemia en LL, pero sí en el 0.6-4.8% de los casos de leucemia linfoblástica aguda. Además, se ha encontrado que la expresión clínica puede estar asociada con una translocación T (17;19), lo que sugiere la inducción de la proteína relacionada con la hormona paratiroidea6.
Se han identificado cuatro tipos de hipercalcemia relacionados con malignidad: hipercalcemia asociada a neoplasias malignas, hipercalcemia osteolítica local, hipercalcemia inducida por 1,25-vitamina D y secreción ectópica de hormona paratiroidea auténtica7-9.
La hipercalcemia asociada a neoplasias malignas representa aproximadamente el 80% de las hipercalcemias relacionadas con malignidad. Se considera como la manifestación de mecanismos moleculares inducidos secundarios a la acción del activador del receptor del factor nuclear kappa B (NF-kB), que actúa como un activador del receptor de NF-kB ligando (RANK-RANKL). De igual manera, la interacción con las interleucinas 1 y 6, el factor de necrosis tumoral alfa, el factor de crecimiento de transformación beta, las prostaglandinas, el calcitriol y la hormona paratiroidea, que se produce de manera ectópica, lleva a la lesión y la expresión clínica de la hipercalcemia6,10.
De acuerdo con lo anterior, el estudio del calcitriol debe incluirse en pacientes oncológicos que presenten hipercalcemia. En el caso de los linfomas, la hipercalcemia generalmente se debe a la 1,25-vitamina D, la cual promueve la diferenciación osteoclástica11 y causa intoxicación por vitamina D9. Considerando los resultados del calcitriol en el paciente del presente reporte, fue posible descartar la 1,25-vitamina D como causa de la hipercalcemia.
Por otro lado, la hipercalcemia osteolítica local representa el 20% de las hipercalcemias de malignidad, y se asocia frecuentemente con metástasis en el esqueleto y leucemia en los niños. En general, este mecanismo ocurre como consecuencia de la destrucción ósea localizada por células cancerosas invasivas que liberan citocinas y factores estimulantes de osteoclastos de forma directa sobre la superficie ósea6.
En cuanto a las adenopatías cervicales, constituyen un motivo frecuente de consulta en pediatría. En la mayoría de los casos son de origen infeccioso12 y en dos terceras partes no se puede aislar ningún agente infeccioso específico13. De los pacientes que recibieron un diagnóstico definitivo, el virus Epstein-Barr, el cáncer y la enfermedad granulomatosa representaron el 8.86%, el 4.69% y el 4.06% de los casos, respectivamente. El LNH es la etiología maligna más frecuente (46% de los casos). Por otro lado, la tuberculosis fue la enfermedad granulomatosa más frecuente (73.4%)13.
Otro aspecto importante es que los pacientes como el del presente caso, con adenopatías, hepatomegalia, nódulos cutáneos y lesiones líticas, se benefician de un estudio de PET, debido a que brinda la posibilidad de combinar un método de imagen tanto anatómico como funcional, que permite identificar y localizar de manera precisa las lesiones que no pueden ser observadas fácilmente con otros estudios de imagen14. La mayor experiencia que se tiene en pediatría con la PET es, de hecho, en el estudio de LNH. Se utiliza principalmente en la estadificación, la cual se ha logrado precisar en el 32-40% de los casos. Además, de acuerdo con los resultados de la PET, se puede evaluar la respuesta al tratamiento, en reestadificación, ya que se evalúan las masas residuales para diferenciar las lesiones activas de las inactivas, e incluso al planear la obtención de una biopsia o la aplicación de radioterapia15.
Por último, es importante describir el estado actual del paciente: a la fecha, se encuentra iniciando la fase de mantenimiento del protocolo BFM90. Antes de iniciar el mantenimiento, se confirmó que en la PET posterior a la inducción no hubiera evidencia de actividad metabólica tumoral. Los resultados de la biopsia de médula ósea no presentaron evidencia de patología maligna y tampoco se observaron blastos en el líquido cefalorraquídeo (con la aplicación de quimioterapia intratecal).
En un ultrasonido de testículo reciente se detectó una disminución de su volumen, lo cual confirmaría la infiltración testicular previa.
El dolor óseo en la edad pediátrica amerita una exploración física minuciosa para no retrasar el diagnóstico oportuno de cáncer y afectar negativamente el pronóstico del paciente. El dolor óseo no es un síntoma normal ni se debe asociar al crecimiento de un niño o adolescente. Cualquier tipo de dolor óseo en un paciente pediátrico debe recibir atención adecuada y estudio minucioso.
La sospecha de cáncer en un paciente debe ser analizada, idealmente, por el médico de primer contacto, y es importante no iniciar tratamientos sin tener un diagnóstico de certeza.
Este caso es un claro ejemplo de cómo el cáncer continúa siendo diagnosticado en estadios avanzados, cuando ya se afectan de manera considerable el pronóstico y la calidad de vida del paciente y de sus familiares. Considerar el cáncer como un diagnóstico diferencial es deber de todo médico para permitir tanto la detección precoz como el tratamiento oportuno, y así disminuir la mortalidad asociada con patología oncológica, la intensidad del tratamiento, las secuelas y los costos asociados.

