











 nueva página del texto (beta)
nueva página del texto (beta) Inglés (pdf)
Inglés (pdf)
 Artículo en XML
Artículo en XML Referencias del artículo
Referencias del artículo
 Enviar artículo por email
Enviar artículo por email Citado por SciELO
Citado por SciELO  Similares en
SciELO
Similares en
SciELO 
 Permalink
PermalinkIntroduction
Coronaviruses (CoVs) have been studied for nearly nine decades. They have long been associated with a wide variety of respiratory, gastrointestinal (GI), neurological, and multisystemic diseases in many animal species, including humans. Human coronaviruses (HCoVs) were identified shortly after the initial description of many respiratory viruses that cause respiratory diseases during the 1960s. By the 1990s, many aspects of the basic molecular biology, epidemiology, and pathogenesis had been explored. It was clear that their mutation and recombination rates made these viruses highly adaptable to changing tropism and transmission through zoonotic events with epidemic and pandemic potential.
CoVs, which belong to the Nidovirales order, are characterized by the production of nested subgenomic mRNAs and many viral proteins involved in proteolytic processing, genome replication, and subgenomic mRNA synthesis that implicate a highly complex viral replication cycle. They are enveloped, plus-strand RNA viruses with the largest known RNA genomes and infect birds and mammals. The Coronavirinae sub-family is divided into four genera, including the alpha- and beta-CoVs. From these genera, seven CoVs that infect humans are known: 229E, NL63, OC43, HKU1, severe acute respiratory syndrome coronavirus (SARS-CoV), Middle East respiratory syndrome coronavirus (MERS-CoV), and SARS-CoV-2. HCoVs have been known to cause enteric or respiratory infections.
In contrast to the four endemic HCoVs (HCoV-229E, HCoV-NL63, HCoV-OC43, HCoV-HKU1), three epidemic HCoVs have emerged in humans in the last two decades, including the SARS-CoV-2 virus that causes the coronavirus disease (COVID-19). The disease rapidly spread worldwide and was declared a pandemic by the World Health Organization in March 2020. This HCoV causes a mild upper respiratory illness or pneumonia. In older patients and those with underlying comorbidities, the infection often results in acute respiratory distress syndrome (ARDS), multi-organ failure, and death. Recent findings have shown that children account for approximately 1-5% of diagnosed COVID-19 cases, in whom COVID-19 disease seems to be less severe. Although approximately 90% of pediatric patients are asymptomatic or present mild or moderate symptoms, up to 6.7% of cases may be severe. The epidemiological and clinical patterns of SARS-CoV-2 infection and COVID-19 in pediatric patients remain unclear, highlighting the urgent need to advance our knowledge on all aspects of the replication of this virus and its interactions with the host. This review aims to provide a brief description of the molecular biology of CoVs and previously identified HCoVs, which have served as the basis to understand the pathogenesis of SARS-CoV-2 and COVID-19.
Historical perspective on CoVs
CoVs have long been associated with a broad spectrum of diseases in animals and humans. Detailed descriptions of this family of viruses have been included in many excellent reviews1-8. They were first reported in 1933, resulting from studies on infectious laryngotracheitis or gasping diseasea lethal respiratory disease of chickenswhich eventually led to the identification and cultivation of the infectious bronchitis virus (IBV) that would become the prototype of this family of viruses9-11. Subsequently, other CoVs were found to cause GI diseases. In the late 1940s and early 1950s, two related murine CoVs were identified: a virus causing encephalomyelitis in mice called JHM (after Professor J.H. Mueller) and the mouse hepatitis virus (MHV)12-14. Although MHV and JHM were initially considered to be potentially useful models for studies on human hepatitis and demyelinating encephalitis, their relationship with these diseases in humans was not demonstrated at the time. However, antibodies that cross-reacted with MHV were found in studies with human serum15. By the early 1960s, many currently known viruses that caused respiratory disease were identified, including adenoviruses, influenza A, B, and C, para-influenza 1, 2, 3, and 4, respiratory syncytial viruses, and rhinoviruses. However, since these agents were isolated and cultured from about one-third of the cases of common colds or related illnesses, other cases were suspected to be caused by additional unidentified viruses that could not be cultured. Cultures from the human embryonic trachea or nasal epithelium led to the isolation of the B814 virus strain, which was unrelated to any known respiratory viruses and was suspected to be a myxovirus because it proved to be ether-labile16. Other strains were later identified in organ cultures (OC), but only two could be successfully grown in the brain of suckling mice, OC 43 and 3817, which would eventually be adapted to grow in cell monolayers. Simultaneously, other agents were isolated from medical students with a common cold and grown as cell cultures, from which the 229E strain was selected and became the prototype strain18. Studies on the antigenic relation, electron microscopy of viral particles, and the cytopathic effect observed in infected cells showed similarity between the IBV, MHV, and the human B814 and 229E strains. Therefore, they were considered a related group of viruses named CoVs because of their morphological appearance resembling the solar corona19. The three decades that followed showed significant advances in the biology of CoVs20-23. CoVs were identified in many animal species of birds and mammals, and their potential for zoonotic transmission between species became apparent. For example, in the case of the known HCoV strains, HCoV-229E originated in bats and was transmitted to humans through alpacas, and the HCoV-OC43 passed from rodents to humans through cattle. Advances in the molecular biology of CoVs also showed high rates of viral genome mutation and recombination, making CoVs highly flexible to adaptation, changing tissue tropism, and hosts.
The viral pathogenic potential ranges from respiratory or GI diseases to hepatitis, encephalomyelitis, vasculitis, coagulopathies, and neurological damage. The diseases severity could often be related to immunopathological aspects of the antiviral response and underlying risk factors. However, since most human infections led to only moderate disease, HCoV was considered more a nuisance than a threat to human health. In 2002, the zoonotic SARS-CoVoriginated in bats and passed to humans by palm civets24led to over 8,000 cases by the end of the pandemic (in June 2003). With a mortality rate of close to 9.5%, it brought more attention to HCoVs. Additional HCoVs were identified shortly after that: HCoV-NL63 in 200425 and HCoV-HUK1 in 200526. As for the endemic HCoV-229E and HCoV-OC43, infections with the more recently identified HCoV-NL63 and HCoV-HUK1 also resulted in mild and self-limiting diseases. However, they were sometimes associated with severe lower respiratory infections in infants, older adults, or immunocompromised patients. In 2012, MERS-CoV emerged27 also as a zoonotic virus that originated in bats, and after a bat to camel switching event may have transferred to humans from dromedary camels. Two MERS-CoV outbreaks caused over 2000 deaths in Saudi Arabia and South Korea, with a mortality rate close to 35%. The elderly and people with underlying morbidities often developed a disease more severe and fatal. The SARS-CoV and MERS-CoV pandemics contributed to recognize the previous knowledge of CoVs potential as zoonotic agents that can rapidly evolve to cause human pandemics28.
Molecular biology of CoVs
Virus structure and genome organization
CoVs are classified into four genera: alphacoronavirus, betacoronavirus, gammacoronavirus, and deltacoronavirus, in the Orthocoronavirinae subfamily, Coronaviridae family, and Nidovirales order. They can infect a wide variety of hosts, including avian, murine, bovine, swine, feline, canine, bats, and humans. HCoVs belong either to the alpha- or betacoronavirus genera. The seven HCoVs known today are the alphacoronaviruses, HCoV-229E and HCoV-NL63, and the betacoronaviruses, HCoV-OC43, HCoV-HKU1, SARS-CoV, MERS-CoV, and SARS-CoV-2 (Table 1).
Table 1 Human coronaviruses
| Coronaviridae | Strains | Receptor | Animal host | Date identified (year) |
|---|---|---|---|---|
| Alpha-coronavirus | HCoV-229E | Human aminopeptidase N (CD13) | Bats | 1966 |
| HCoV-NL63 | ACE2 | Palm civets, Bats | 2004 | |
| Beta-coronavirus | HCoV-OC43 | 9-O-Acetylated sialic acid | Cattle | 1967 |
| HcoV-HKU1 | 9-O-Acetylated sialic acid | Mice | 2005 | |
| SARS-CoV | ACE2 | Palm civets, Bats | 2003 | |
| MERS-CoV | DPP4 | Bats, Camels | 2012 | |
| SARS-CoV2 | ACE2 | Bats | 2019 |
The virions are spherical or pleomorphic enveloped particles with a diameter of 80-120 nm, formed by four or five structural proteins: the spike glycoprotein (S), the hemagglutinin-esterase (HE, present in some betacoronaviruses), the membrane glycoprotein (M), the envelope glycoprotein (E), and the nucleocapsid protein (N). The S glycoprotein is a large type I transmembrane polypeptide with an N-terminal exodomain and a C-terminal endodomain. The S protein assembles into trimeric spikes that protrude from the virus surface with a club-like appearance and mediates receptor binding and membrane fusion. The HE forms shorter dimeric projections and may participate during cell entry and egress. The M protein is the most abundant virion component and is embedded through three transmembrane domains supporting the viral envelope.
In contrast, the E protein is a small transmembrane protein present in lower copy numbers. The viral envelope surrounds a nucleocapsid with helical symmetry, which is not characteristic for positive-strand RNA viruses but rather typical of negative-strand RNA viruses. The helical nucleocapsid is formed by the N phosphoprotein, which associates with the RNA genome in a beads-on-a-string fashion8.
CoVs possess the largest known genomes of RNA viruses, ranging from 27 to 31 kb. Their genome is a non-segmented, single-stranded RNA of positive polarity, modified with a 5-cap, and 3-poly(A) tail that can be translated once it enters the cells cytoplasm. Indeed, the coronavirus genome is infectious when transfected into permissive host cells. The extensive coronavirus genome encodes multiple polypeptides and is organized in a highly conserved 5-replicase/transcriptase-S-E-M-N-3 gene arrangement, with various smaller ORFs interspersed among the S-E-M-N structural genes. The genes that code for the structural proteins occupy less than one-third of the genome at the 3-end. The remaining two-thirds are occupied by a single gene that codes for the viral replicase/transcriptase proteins. Like other single-stranded RNA viruses, the coronavirus genome has a high mutation rate of about 10−4 nucleotide substitution/site/year due to an inefficient proof-reading mechanism8. Furthermore, CoVs may adapt rapidly to changing ecological niches due to high recombination frequencies that originate from the complex mechanisms responsible for synthesizing various species of viral RNA during genome transcription and replication (Fig. 1).
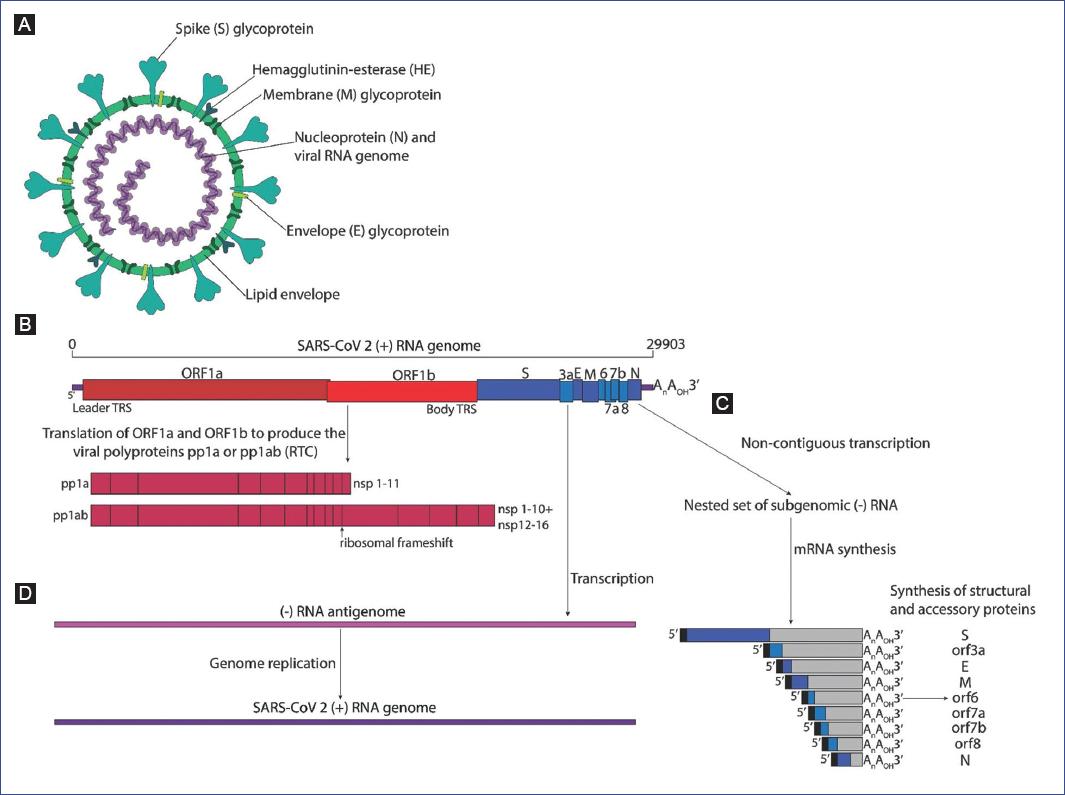
Figure 1 Diagram of the coronavirus virion, genome organization, polyprotein processing, subgenomic RNA production, and genome replication. A: The virion consists of the helical nucleocapsid formed by the N protein-associated RNA genome, surrounded by a lipid envelope where the M, S, E (and HE in some betacoronaviruses) transmembrane glycoproteins are embedded. B: The positive-strand RNA genome is organized with the Orf1a and Orf1b occupying nearly two thirds of the 5 end of the genome, which are translated to produce the autoproteolitically processed pp1a and pp1ab polyproteins, respectively, yielding nsp1 to nsp16 (shown in different shades of red). The remaining one third 3 end of the genome encodes the S, E, M, N structural and the accessory proteins (shown in different shades of blue). C: The mRNAs that encode the structural and accessory proteins are produced through the non-contiguousnestedtranscription of negative-strand subgenomic RNAs that direct the synthesis of their corresponding complementary mRNAs, which share common 5 and 3 ends. Only the 5 most ORF is translated from each nested mRNA (shown in boxes with different shades of blue). D: The full-length positive strand RNA genome is transcribed to produce a full-length negative-strand antigenomic RNA that serves as the template for the synthesis of the positive-strand RNA during genome replication.
Coronavirus replication cycle
Attachment and entry
Coronavirus binding and entry into the host-cell depend on the S glycoprotein, which is the principal determinant of host species range and tissue tropism. A variety of studies performed through the 1990s demonstrated that swapping of the cellular receptor or the viral S protein is sufficient to redirect viral tropism and alter the degree of virulence7. The S protein comprises two domains: S1 and S2. The interaction between the highly variable S1-RBD domain (receptor binding domain) and the cellular receptor induces a conformational change that promotes membrane fusion between the viral and cellular membranes through the conserved S2 domain. Cell receptors for many CoVs have been identified and, like the S protein, are substantial determinants of pathogenicity, tissue tropism, and host range. The carcinoembryonic antigen-related cell adhesion molecule 1 is a receptor for MHV, whereas the 9-O-acetylated sialic acid is the receptor for BCoV, HCoV-OC43, and HCoV-HKU1. The aminopeptidase-N is the receptor for the transmissible gastroenteritis virus, the porcine respiratory coronavirus, and HCoV 229E. The dipeptidyl peptidase-4 is MERS-CoV and the angiotensin-converting enzyme 2 (ACE2) is SARS-CoV, HCoV-NL63, and SARS-CoV-2. CoVs enter through cell endocytosis or at the plasma membrane, where other proteases, like furin, the transmembrane protease serine 2 (TMPRSS2), or the airway trypsin-like protease TMPRSS11D may participate in S1/S2 cleavage, facilitating membrane fusion, as in the case of HCoV-229E, SARS-CoV, and SARS-CoV-2 infection20,21.
Viral genome expression and replication
Upon fusion of the viral envelope membrane to either the plasma or endosome membrane, the viral nucleocapsid is released into the cytoplasm. Subsequently, cellular ribosomes translate the viral genome through a cap-dependent mechanism to synthesize two large polyproteins, pp1a and pp1ab. These polyproteins are encoded in overlapping open reading frames ORF1a and ORF1b that are translated through a ribosomal frameshift mechanism in which the ribosome shifts one nucleotide in the -1 reading frame of ORF1a into the ORF1b frame. The resulting pp1a and pp1ab are cotranslationally and autoproteolytically processed to produce 15 or 16 nonstructural proteins (nsp) (Table 2). Nsp1 to nsp11 are generated from pp1a and nsp12 to nsp16 from pp1ab. Nsp encoded in the ORF1a and 1ab genes form the replicase/transcriptase complex (RTC) that transcribes the full-length positive-strand genomic RNA to direct the synthesis of a full-length negative-strand RNA that functions as a template for the synthesis of new genomic RNA. The RTC assembles in a reticulovesicular network of modified ER membrane induced in the infected cell, where viral RNA replication takes place. The new genomic RNA molecules are encapsidated and incorporated into progeny virions on membranes of the endoplasmic reticulum-Golgi intermediate compartment (ERGIC)20-22.
Table 2 Functions of coronavirus nonstructural proteins
| Protein | Functions |
|---|---|
| Nsp1 | Host mRNA degradation; translation inhibition; cell cycle arrest; inhibition of IFN signaling |
| Nsp2 | Unknown |
| Nsp3 | Papain-like proteases (PL1pro, PL2pro); poly(ADP-ribose) binding; DMV formation; IFN antagonist; nucleic acid binding; deubiquitinating activity |
| Nsp4 | DMV formation |
| Nsp5 | Main protease (Mpro, 3CLpro) |
| Nsp6 | DMV formation |
| Nsp7 | Single-stranded RNA binding |
| Nsp8 | Primase |
| Nsp9 | Part of replicase complex |
| Nsp10 | Part of replicase complex |
| Nsp11 | Unknown |
| Nsp12 | RNA-dependent RNA polymerase |
| Nsp13 | Helicase; RNA 5'-triphosphatase activity |
| Nsp14 | 3' - 5' exoribonuclease; (guanine-N7)- methyltransferase |
| Nsp15 | Endonuclease |
| Nsp16 | 2'O-methytransferase |
DMV: double-membrane vesicle; IFN: interferon; RNA: ribonucleic acid.
Polyprotein processing
As in other positive-strand RNA viruses, coronavirus polyproteins proteolytic cleavage is a crucial process for regulated viral gene expression. However, in CoVs, polyprotein processing implicates multiple cleavage sites that produce many enzymes involved in coronavirus RNA synthesis and modification that is unique in RNA viruses. Such complex proteolytic processing depends on the main protease (Mpro)a chymotrypsin-like (picornavirus 3C-like) cysteine protease (3CLpro) that resides in nsp5 and cleaves at 11 sites to generate 13 polypeptides (nsp4-nsp16)and on one or two accessory papain-like cysteine proteases (PL1pro and PL2pro) that reside in nsp3 and process nsp1-nsp4, the C-terminus of nsp4 being cleaved by the Mpro.
Some of the 16 nsp functions produced after polyprotein cleavage are summarized in table 2. These include the core viral enzymes responsible for RNA synthesis during viral genome transcription and replication: the RNA-dependent RNA polymerase (nsp12), the RNA polymerase primase (nsp8), the RNA helicase (nsp13), and the 3-5 exoribonuclease (nsp14), as well as other RNA processing enzymes, such as poly(U) endoribonuclease, cyclic phosphodiesterase, and adenosine diphosphate-ribose-phosphatase. Additional enzymes include those required for 5 capping of viral RNAs through the (guanine-N7)-methyltransferase activity of the bifunctional nsp14 and nsp16 S-adenosyl-methionine-dependent RNA (nucleoside-2O)-methyltransferase (2O-MTase) activity, which may function together with nsp1529.
Viral genome transcription
After production and processing of viral polyproteins, the RTC is assembled to direct transcription of the genes that occupy the remaining one-third of the genome near the 3-end that is not directly translated from the positive-strand RNA genome and encodes the structural proteins (S-E-M-N), and 1 to 8 additional accessory proteins20. These genes are transcribed through a mechanism unique to the Nidovirales order, involving non-contiguous transcription of the 3-end of the viral genomewhere the viral RNA-dependent RNA polymerase (RdRp) skips from one part of the genome to the next to generate a nested set of subgenomic negative-strand RNAs. The subgenomic RNAs contain common 3-ends and a common leader encoded at the 5-end of the genomic RNA. Transcription termination and acquisition of the leader 5 RNA occur at transcription regulatory sequences located between ORFs. These negative-strand subgenomic RNAs serve as templates for synthesizing subgenomic mRNAs with common 5 leader and 3 sequences. Translation of these mRNAs produces proteins only from the ORF closest to the 5 leader on each of the nested mRNAs8.
Replication compartments
RTC associates with ER-derived membranes (remodeled by nsp3, nsp4, and nsp6) to form a pervasive network of convoluted membranes (CM), double-membrane vesicles (DMV), and vesicle packets (VP), where viral proteins and RNA are compartmentalized. Electron tomography revealed that the membrane network is continuous with the ER and contains large vesicles with abundant viral dsRNA and that they are not connected to the cytoplasm. Analysis at late stages of infection suggests that the membrane network involved in virus replication may merge with membranes where virus assembly occurs30. These virus-induced membrane compartments represent replication organelles (RO), where viral genome transcription and replication seem to be orchestrated. The nsp3, nsp4, and nsp6 are transmembrane proteins that anchor the RTC to induce the formation of structures that provide a scaffold responsible for viral RNA synthesis and protection of the RNA intermediates from cellular antiviral mechanisms8. The crucial steps of virus subgenomic and genomic RNA and mRNA synthesis are controlled at these sites and implicate dynamic interactions of nsps, viral RNAs, and the N protein. The subgenomic RNA (produced at these sites) directs the synthesis of structural proteins, while the recruitment of N protein oligomers through association with nsp3 may promote the assembly of ribonucleoprotein complexes31.
Virus assembly
The S, M, HE, and E proteins are synthesized in association with the ER. These proteins transit to the ERGIC, where nucleocapsids assembled by the N protein and newly produced genomic RNAs are encountered. Virion assembly proceeds through interactions among the structural proteins, where the M protein plays a central role. Interestingly the M protein and the minor E protein were shown to be necessary and sufficient for the formation of virus-like particles (VLPs). VLPs, devoid of S spikes, are morphologically similar to virions and are released from cells through the same pathway as complete virions. During infection, the interaction between M and N proteins directs nucleocapsid and membrane components assembly when packaging signal sequences selectively package the genomic RNA, leading to the formation of virions transported to the plasma membrane and released by exocytosis7 (Fig. 2).
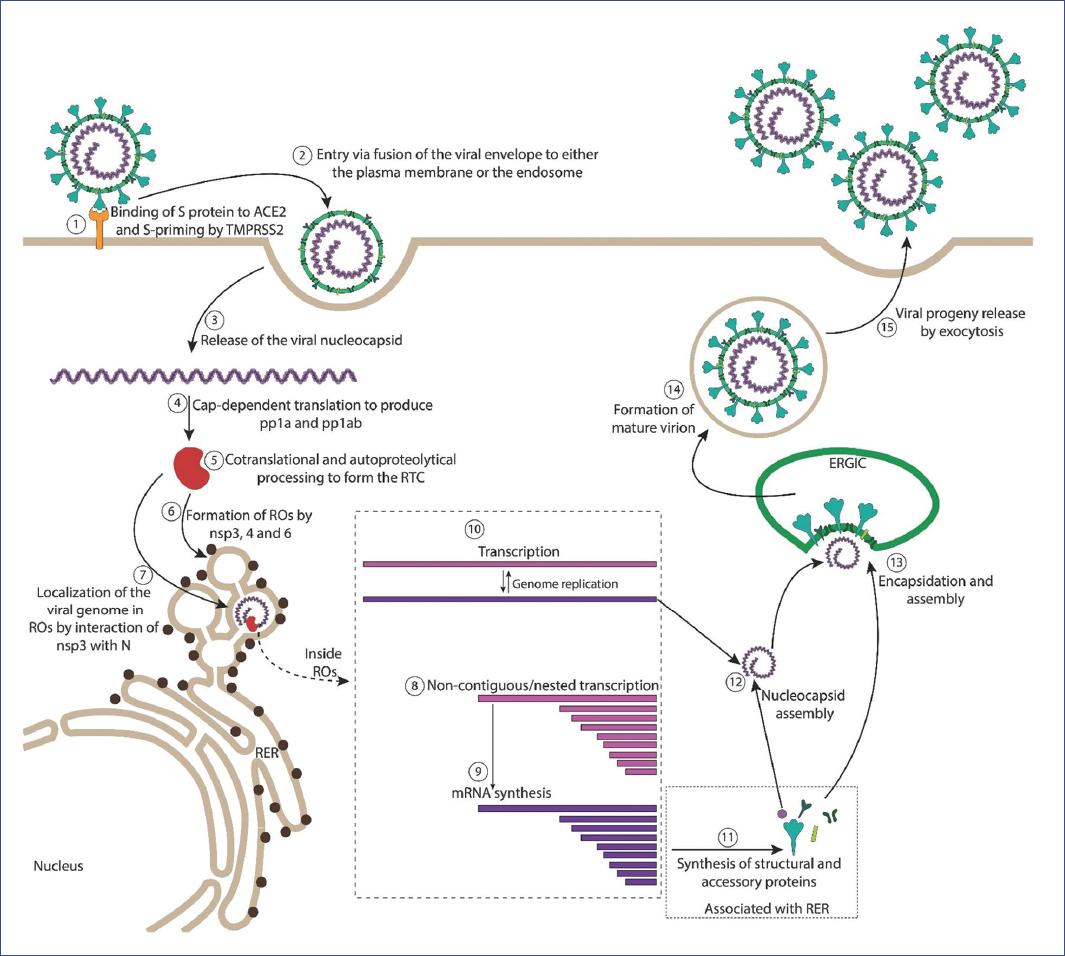
Figure 2 Coronavirus replication cycle. (1) The coronavirus virion binds to the cellular receptor and (2) enters the cell through fusion of the viral envelope either with the plasma membrane or the endosome. (3) The nucleocapsid is released into the cytoplasm, where (4) cellular ribosomes translate the 5 end of the genome through a cap-dependent mechanism to produce the pp1a and pp1ab polyproteins that are (5) cotranslationally autoproteolytically processed that yield the nonstructural proteins (nsps 1-16) that form the replicase/transcriptase complex (RTC), and other proteins that alter many cell functions. (6) The nsp3, 4, and 6 participate in formation of virus-induced replication organelles (ROs), where (7) the viral genome localizes and (8) non-contiguous transcription of the 3 end of the genome produces the nested subgenomic RNAs that are (9) transcribed into the mRNAs that encode the viral structural S, E, M, N proteins. (10) The full-length positive-strand RNA is also transcribed in these sites producing the full-length negative-strand RNA that serves as the template for viral genome replication. (11) The mRNAs that encode the structural proteins are translated by ribosomes that associate with the ER and are translocated, glycosylated, and processed through ER and Golgi. (12) Association of the N protein with newly synthesized viral RNA genomes forms the nucleocapsid that interacts with the (13) ERGIC membranes acquiring the S, M, E proteins, leading to (14) formation of the mature virion that is released from the cell (15) through exocytic vesicles.
Virus-host interactions
Virus-induced alteration of the infected host cell
Viruses are obligate intracellular parasites that depend on the cellular architecture and functions for their replication. Upon infection, viruses may induce extensive cell structure reorganization resulting in cytopathic effects due to cell membrane or cytoskeleton components alterations. In addition, viruses may target several physiological and biochemical aspects of cells by inducing changes in signaling pathways, cellular gene expression, regulation of synthesis of cellular macromolecules, and alterations in the overall cellular metabolism, resulting in enhanced viral replication32,33.
The concentration and compartmentalization of macromolecules needed for viral genome replication and gene expression are induced during viral replication, resulting in specialized virus-induced cellular microenvironments termed viral factories, viroplasms, replication centers, compartments, or organelles. Such RO also represents a physical barrier that protects the viral genome from cellular defenses34-37.
As mentioned before, CoV-replication is accompanied by a variety of intracellular membrane rearrangements derived from the ER, resulting in the formation of ROs that display various discernible morphologies such as DMVs, which range from 150 to 350 nm in diameter, and are sites where viral dsRNA replication intermediates accumulate. DMV may fuse to form larger structures called VP. DMVs are interconnected with convoluted reticular membranes (CMs), which range from 0.2 to 2 mm in diameter and are the primary sites where viral replicase proteins are colocalized. At late times post-infection, structures known as large virion-containing vesicles are formed, enriched with viral structural proteins38-40. The nsp3, nsp4, and nsp6 are integral membrane replicase proteins that promote ROs assembly by inducing membrane remodeling and recruitment of factors necessary for viral genome transcription and replication. The sole expression of nsp3 and nsp4 is sufficient to induce membrane curvature41. Nsp3 is a large multifunctional protein comprising up to sixteen different domains thought to function as a scaffold for RO assembly and regulation of RO-associated activities through its interaction with several proteins that participate in replicating and transcribing the viral genome. For example, the ubiquitin-like (ubl) domain of nsp3 is involved in ssRNA-binding and interaction with the viral nucleocapsid N protein. Interaction of nsp3 with N tethers the viral RNA to the RTC in ROs early during infection, and deletions within the ubl domain result in impaired viral RNA replication42. Several other viral proteins are known to be part of ROs, including the nsp2, 5, 7-10, and nsp12-16 and the N protein, together with cellular proteins that participate in vesicular trafficking, ubiquitin-dependent, and autophagy-related proteins and translation factors that localize at the cytoplasmic face of ROs43.
Innate antiviral response
It was mentioned earlier that ROs are thought to shield the viral RNA and proteins from recognition by the host antiviral mechanisms, as viral RNA and replicase subunits become sensitive to nuclease or protease treatment only after membrane disruption by non-ionic detergents44. Therefore, the formation of ROs is a crucial mechanism for evasion of the cellular antiviral response. Although the specific antiviral mechanisms altered or inhibited by SARS-CoV-2 through reorganization of intracellular membranes and redistribution of cellular components have not been studied in detail, evidence indicates that some CoV proteins induce intracellular stress pathways, which are known to crosstalk with the immune response45. Since HCoV-replication takes place in ROs that are assembled by remodeling of ER membranes, it is expected that the alterations of the ER lead to the activation of the unfolded protein response (UPR). This stress-induced signaling pathway leads to inhibition of translation while increasing the synthesis of ER membranes and chaperones needed to counteract the accumulation of misfolded or unfolded proteins. Expression of the viral structural S protein correlates with transcriptional activation of the glucose-regulated protein 78 and 94 (GRP78/94) and upregulation of the PKR-like ER kinase (PERK) pathway from the UPR and induce the innate immune response by inducing the expression of the chemokine CXCL246,47. The viral accessory protein ORF9a is a transmembrane protein that localizes to the ER and is also a modulator of the ER stress response, modulating the PERK pathway and resulting in the phosphorylation, ubiquitination, and degradation of the interferon (IFN)-alpha receptor subunit 148. ORF8b forms intracellular aggregates, induces ER and lysosomal stress, and causes cell death in epithelial cells. This viral protein induces the activation of the intracellular sensor NOD-, LRR-, and pyrin domain-containing protein 3 (NLRP3) and release of the pro-inflammatory cytokine IL-1b, which may trigger aberrant activation of pro-inflammatory monocyte-macrophages49. The activation of the PERK pathway subsequently leads to phosphorylation of the translation-initiation factor eIF2a and, therefore, translation shutoff50.
More than 7 months have elapsed since the discovery of SARS-CoV-2, which may seem a long time for the lockdown, but represents a relatively short time for an exhaustive study of the immune responses development and evolution against this virus in a human population. As a result, many of the studies reported to date are either clinical case studies or studies with small groups of patients that have presented severe symptoms or symptoms considered atypical. However, some key aspects of cellular antiviral mechanisms and viral countermeasures have been described. Given the similarity between SARS-CoV-2 and SARS-CoV and the conserved mechanisms of the innate antiviral response, detection of SARS-CoV-2 by intracellular receptors is likely mediated by Toll-like receptors and RIG-I-like receptors. These receptors trigger downstream signaling and activation of transcription factors, such as IFN-regulatory factors (IRFs) and nuclear factor kappa B (NFkB), which result in the expression of IFN response genes and other cytokine genes51,52.
The genome of SARS-CoV-2 also encodes proteins that counteract the antiviral activity of specific intracellular immune receptors. The structural N protein of SARS-CoV-2 inhibits the type I IFN response53. It is also suggested that the nonstructural viral protein nsp3 is a type I IFN antagonist20 that promotes RO assembly. Nsp9 targets MIB1, an E3 ubiquitin ligase that promotes TNF-induced apoptosis. Nsp13 targets TBK1, TBKBP1, and the NFkB pathway.
Moreover, the nsp15 endonuclease (EndoU) that is highly conserved among CoVs targets RNF41 (an E3 ubiquitin-ligase for MYD88) and prevents activation of MDA5, OAS/RNase L, and PKR54,55. In addition, viral accessory proteins can block innate immune factors: ORF9b targets TOMM70, the mitochondrial import receptor that mediates activation of IRF3 in mitochondria56. ORF9c targets NLRX1 (a negative regulator of IFN-I), F2RL1 (modulator of the inflammatory responses and innate and adaptive immunity), and NDFIP2 (a factor that limits cytokine signaling by promoting degradation of JAK1 by NEDD4-mediated ubiquitination). ORF3a targets TRIM59 (a regulator for innate signaling pathway), and the ORF6 protein blocks the IFN-inducible mRNA nuclear export complex NUP98-RAE1, therefore altering the transport between the nucleus and the cytoplasm57. All of these SARS-CoV-2 cellular protein-protein interactions lead to an impaired antiviral response, with inefficient production of type I IFN and increased cell death, which is linked to severe outcomes of COVID-19 that results from hyper inflammation and the cytokine storm51,58. In contrast to mild HCoVs infection22, serum samples from severe COVID-19 patients are characterized by low levels of IFN and elevated levels of chemokines and pro-inflammatory cytokines that result in peripherally derived macrophages in the lungs and an influx of activated neutrophils59,60.
Adaptive immune response
There is increasing evidence suggesting that exposure to SARS-CoV-2 infection generates a good adaptive immune response, both cellular and humoral, which are encouraging results for a good prognosis for protection against reinfection and vaccine development. However, this response appears to be more efficient for patients with severe COVID-19 than for asymptomatic or mildly symptomatic patients61.
Together with IgMs, immunoglobulins A in mucosal membranes are the first antibodies generated against invading agents and can be observed since the first week of infection. IgGs against SARS-CoV-2 are usually detected between 10 days and 2 weeks after the onset of symptoms. These immunoglobulins and complement proteins are produced at expected levels to confer protection; however, their duration has yet to be determined62.
Other findings suggest that SARS-CoV-2 could evade immune surveillance through a hidden RBD of the spike protein, which is not well recognized by immune factors compared to an exposed RBD63. Although virus-cell recognition may be generally established through the interaction between viral structural proteins and cell surface receptors, cells that do not express virus receptors may be infected by antibody-dependent enhancement (ADE). It has been suggested that ADE could be promoted by the presence of non-neutralizing or sub-neutralizing antibodies against the S-protein64. Similar to what has been reported for SARS-CoV, this mechanism could aid SARS-CoV-2 to infect immune cells lacking ACE2 expression by binding to Fcg receptors for cell entry65. Nonetheless, whether SARS-CoV-2-infection can be mediated by ADE or infect immune cells remains unknown66.
In addition to humoral immunity, protection against different pathogens is mediated by T-lymphocytes, which may confer long-term immunity67. A T-cell response is more likely to be induced by the nsp. However, there is evidence that the S protein can also induce memory T-cell differentiation, as measured by S-RBD-specific T cell production of IFNg68. CD4+ cells mainly differentiate toward memory Th1 and Th17 helper cells, and there is also evidence for the development of cytotoxic CD8+ cells against SARS-CoV-2 infected cells60. Some studies have shown that the number and activity of CD8+ cells are higher than the response of CD4+ cells69,70.
A good observation has been discovering cross-reactivity both for cellular and humoral responses, possibly with other endemic HCoVs or with SARS-CoV, which is likely because nsps of SARS-CoV-2 share high sequence identity with other HCoVs. A recent report has shown the presence of CD4+ T cells that recognize a panel of SARS-CoV-2 specific peptides in 40-60% of unexposed individuals (samples collected between 2015 and 2018)71. In addition, cross-immunity has also been observed with humoral response against the S protein, although most of the cross-reactive antibodies are non-neutralizing72.
An interesting observation is the low incidence of severe COVID-19 cases in children. Analysis of human memory B cells (MBC) at different ages has shown that CD27dull and CD27bright represent sequential MBC-developmental stages, leading to the proposal that CD27dull MBCs can expand and differentiate in response to new antigens. Based on these findings, Carsetti et al. proposed that the protective action of non-antigen specific natural antibodies (NAbs) produced by CD27dull MBCs, which are more abundant in children than in adults, could be a determining factor; however, this hypothesis remains to be confirmed73,74.
Severe COVID-19 patients may develop lymphopenia (with depletion of CD4+ and CD8+ cells), which could be induced through signaling exhaustion through inactivation of cytotoxic lymphocytes by IL-6 and IL-8 or viral-mediated apoptosis. In Wuhan, RNA obtained from bronchoalveolar lavage fluids (BALF) and peripheral blood mononuclear cells (PBMC) from SARS-CoV-2 patients indicated an increase in the expression of pro-apoptotic genes and p53-targets, suggesting that lymphopenia may be caused by activation of the p53 signaling pathway and induction of apoptosis in lymphocytes75. One of the viral components responsible for apoptosis induction could be the accessory protein ORF3a, which is known to induce apoptosis76. Recently, it was shown that cells expressing SARS-CoV-2 ORF3a displayed increased annexin V and propidium iodide cell staining, as well as increased activation of caspases, cytochrome C release from mitochondria, and other apoptotic markers77.
Pathogenesis
Clinical manifestations
As previously described above, infection with the SARS-CoV-2 results in a respiratory illness that has been named COVID-19. Most infected people are either asymptomatic or manifest mild symptoms such as fever, headaches, cough, dyspnea, loss of taste and smell, and myalgia or fatigue78. The average person takes 5-6 days after infection for symptoms to show, but in some rare cases, symptoms appear after up to 14 days79. In more severe cases, the disease causes pneumonia and ARDS. The global infection fatality rate (IFR) is estimated to be around 0.68%, according to a recent meta-analysis80, but the specific IFR varies depending on the country. Males are predominantly affected and represent about 60% of the total death cases due to COVID-19. People with comorbidities such as obesity, diabetes, and hypertension also have a higher probability of severe disease or a lethal outcome. After 7 months since the start of the pandemic, with the rapid spread of the virus and increased patients with severe cases, it has become evident to clinicians that COVID-19 also has several extrapulmonary manifestations. Despite a seemingly protective effect against COVID-19, rare symptoms in pediatric patients have emerged in the case of children. It is now apparent that COVID-19 is not just a pulmonary disease but one with pulmonary-hematological-endothelial-inflammatory consequences.
RESPIRATORY
SARS-CoV-2 is transmitted through respiratory droplets and aerosols, direct or indirect respiratory-tract exposure, and potentially by the fecal-oral route81,82. It has a high tropism for the respiratory tract epithelial cells where there is increased expression of its entry receptor, ACE2, including alveolar epithelial type II cells in the lung parenchyma83. Viral replication is understood to first occur in the upper respiratory tract and then further infect and replicate in the lower respiratory tract. In contrast to SARS-CoV, which infects mainly the lower respiratory tract, SARS-CoV-2 can activate replication in the upper respiratory tissues84, explaining the virus continuous high pharyngeal shedding, even in asymptomatic cases61, resulting in the more efficient transmission of SARS-CoV-2 compared to SARS-CoV. Asymptomatic and pre-symptomatic transmission is estimated to account for around half of all cases of COVID-1985.
SARS-CoV-2 replication in the lungs can manifest as ARDS in severe cases of COVID-1978. ARDS is a life-threatening lung condition that prevents oxygen from getting to the lungs and into the circulation in the bloodstream, resulting in death or acute lung injury. This later stage in severe cases of COVID-19 resembles SARS-CoV and MERS-CoV infections in terms of viral replication in the lower respiratory tract86,87. The disease can cause a secondary viremia, followed by an extensive infection of organs that express the viral entry receptor ACE2 such as the heart, kidney, GI tract, and vast distal vasculature88-90. The spread of the virus, which occurs on average around week 2 after disease onset, correlates with clinical deterioration. However, the exacerbation of the disease resulting in organ dysfunction and death is attributed to direct viral organ damage and a consequence of immune-mediated injury induced by SARS-CoV-288. Two distinctive features have been observed in severe and critical patients with COVID-19: a progressive increase of inflammation and a notable hypercoagulation trend.
GASTROINTESTINAL
One of the most commonly reported extrapulmonary manifestations of COVID-19 are GI symptoms, which have been associated with a longer duration of illness, but no association with increased mortality91. The most common symptoms are anorexia, nausea, vomiting, diarrhea, and abdominal pain91,92. Symptoms such as vomiting and diarrhea were often reported during the SARS-CoV and MERS-CoV outbreaks, and more frequently than for COVID-1993-96. Most COVID-19 patients show GI symptoms after respiratory symptoms, although they have also been reported earlier but less frequently95. Intestinal glandular cells express the ACE2 receptor; thus, virus-mediated direct damage is a possibility. The SARS-CoV-2 nucleocapsid protein has been found in gastric, duodenal, and rectal epithelial cells, as well as in glandular enterocytes97. Furthermore, viral RNA shedding in stool has been reported to occur during infection. In some cases, it is detected several days after symptom resolution; therefore, it is thought to be a possible source of viral transmission81,97. In COVID-19 patients, evidence suggests microvascular small-bowel injury and evidence supporting inflammation-mediated tissue damage in the stomach, duodenum, and rectum91,97.
NEUROLOGICAL
The most common symptoms linked to neurological damage in mild cases of COVID-19 are headache, dizziness, myalgia, fatigue, anorexia, anosmia, and ageusia. Nasal epithelial cells have the highest expression levels of ACE2 in the respiratory tract83,98, which could explain the reported loss of smell and taste in many infected people. Severe neurological manifestations are not as common as other extrapulmonary manifestations of COVID-19; however, reports are rapidly increasing. In more severe cases, symptoms such as confusion, impaired consciousness, and acute strokes can occur99,100. A few patients have developed Guillain-Barré syndrome101,102, meningoencephalitis103,104, hemorrhagic posterior reversible encephalopathy syndrome105, and acute necrotizing encephalopathy (which is related to intracranial cytokine storm) that included the brainstem and basal ganglia104,106. In a few of these COVID-19 patients, the virus has been detected in the cerebrospinal fluid. SARS-CoV and MERS-CoV (and other CoVs) are known to have neuroinvasive potential since they can spread from the respiratory tract to the central nervous system (CNS)107-109. It is proposed that the virus could access the brain through circulation or an upper nasal transcribrial route110. This neuroinvasive capability of SARS-CoV-2 could damage the CNS by misdirecting the host immune responses, which could be associated with autoimmunity and viral replication induced damage to CNS cells.
COAGULOPATHIES
At the beginning of the SARS-CoV-2 outbreak, most of the lung damage seen initially in COVID-19 patients was thought to be due to acute viral pneumonia. However, thrombotic complications and severe inflammation have been reported in critical COVID-19 patients. The first reports of clotting disorders were observed in February in China, where the frequent observation of thrombocytopenia, prolonged thrombin time, and elevated D-dimer levels was reported in severe COVID-19 patients111,112. The data suggested disseminated intravascular coagulation (DIC) or pre-DIC. Studies from the Netherlands and France later showed a 30% incidence of thrombotic complications in intensive care unit patients with COVID-19. Among the observed thrombotic complications, pulmonary embolism was the most frequent (80%); however, deep-vein thrombosis, ischemic stroke, myocardial infarction, and systemic arterial embolism were also reported113,114. Systemic anticoagulation (AC) treatment has been used in critical patients to improve their outcomes. However, a debate over the appropriate dosage (standard vs. high doses) of AC is still ongoing. Therefore, clinical trials like IMPROVE-COVID from Columbia University have started to assess the effectiveness of AC treatments and their dosage115. Coagulation disorders have also been previously reported for several SARS-CoV cases; thrombocytopenia was also frequent in MERS-CoV patients, although data are less available116. Interestingly, SARS-CoV has been shown to upregulate procoagulant genes and genes associated with the coagulation pathway in vitro117,118. SARS-CoV nucleocapsid protein has also been shown to induce the human fibrinogen-like protein-2 prothrombinase gene through activation of the transcription factor C/EBP-a119. Furthermore, transgenic mouse models infected with MERS-CoV activate coagulation cascades and form microthrombi in pulmonary vasculature120.
Clot formation is thought to occur for three reasons:
1) Due to the direct infection of endothelial cells by SARS-CoV-2. Endothelial cells express ACE2, and both SARS-CoV-2 and SARS-CoV-2 can infect engineered human blood vessel organoids in vitro and have been detected in vascular beds of different organs in patients with COVID-1990,121,122. Viral replication can cause alveolar endothelial dysfunction, platelet activation, generation of neutrophil-platelet aggregates, neutrophil migration, and fibrin and microthrombus formation. When uncontrolled, these alterations would trigger secondary fibrinolysis, coagulation factors depletion, and consequently DIC and diffuse alveolar hemorrhage112.
2) Inflammation during severe cases of COVID-19 can promote coagulation. Recruitment of immune cells by a direct viral infection of endothelial cells or by pro-inflammatory signaling can cause widespread endothelial dysfunction, inflammation, and a procoagulant state. Accumulation of inflammatory cells within the endothelium and cell death has been observed in severe COVID-19 patients, suggesting endotheliitis in several organs as a consequence of SARS-CoV-290. Complement components in the lung and skin, colocalizing with SARS-CoV-2 proteins, have also been reported, suggesting complement-mediated thrombotic microvascular injury syndrome123.
3) Other factors not specific to SARS-CoV-2 infection could also be involved, such as patients with high clotting risk factors, like in older adults; obese or overweight patients, and those with diabetes or high blood pressure. A state of hypoxia and long periods of immobilization in critical patients could also contribute to clotting.
COVID-19 in children
Pediatric COVID-19 cases have been estimated to account for only 1-5% of the confirmed cases124. The severity and the mortality of the disease among children is significantly lower125, causing only mild symptoms such as fever and coughing in most cases and dyspnea in a few cases. Neonates have a nonspecific presentation with fever and lethargy. Most hospitalized children with severe COVID-19 have been associated with other underlying conditions124. Therefore, it has been suggested that age has a possible protective effect, one which may hold the key to find therapeutic targets. Two main theories that could operate in combination are thought to explain the protective effect of age: (1) the differences in pediatric immune responses compared to adults74, and (2) the differences in the availability of viral binding sites necessary for viral entry in airway epithelial cells126,127. As previously discussed, extensive lung damage and other extrapulmonary manifestations of severe COVID-19 that complicate adult cases are thought to be a consequence of an overactive immune response and not directly related to viral replication. In comparison, childrens immune system is thought to respond less aggressively to SARS-CoV-2 infection. An imbalance in the production of pro- versus anti-inflammatory cytokines may contribute to this process. An example would be the declining production of IL-10 levels with age, which plays an anti-inflammatory role as it decreases macrophage activation and the release of inflammatory cytokines such as IL-6, TNF-a, and IL-1b. Murine models for lung injury have shown a larger increase in neutrophil infiltration and IL-1b levels in adult mice which have lower levels of IL-10 and IL-13 but not in young mice128,129. Furthermore, it has been proposed that childrens IgM B cells (MBCs) can produce NAbs rapidly and abundantly before producing high-affinity IgG antibodies, which would help during the early phases of infection130,131. In this context, since CD27dull MBCs are proposed to respond to new antigens, conferring children a highly adaptable response to new antigens, childrens NAbs would have broader reactivity as they have not yet been selected for reaction to common environmental pathogens74. Another immune difference between children and adults is CD8+ and CD4+ lymphocyte levels. Children before 3 years of age have higher CD4+ cells than adults, and levels of CD8+ cells are slightly increased in elderly adults132. Lymphocytopenia has been reported frequently as a risk factor for most adult COVID-19 patients. It has been proposed that differences in the CD4+/CD8+ ratios between children and adults have a protective effect on children, but it has not been confirmed yet127. Differences between children and adults have also been observed in the availability of SARS-CoV-2 cellular receptors (ACE2 and TMPRSS2) in lung epithelial cells. Transcriptomic data from lung cells across the age span of 30 weeks, 3 years, and 30 years show an increase in the proportion of alveolar epithelial cells expressing ACE2 and TMPRSS2 in adult lungs compared with young lungs133. Furthermore, cell-specific expression analysis of viral entry mediators found that ACE2 and TMPRSS2 expression in airway epithelial and alveolar type 2 (AT2) cells increases with age, with deficient expression in infants and young children134. These data may suggest less available viral entry points in the lung epithelial cells in children compared to adults. Furthermore, a recent Gene Set Enrichment Analysis showed that high expression of ACE2 was also related to the activation of neutrophils, NK cells, Th17 cells, Th2 cells, Th1 cells, dendritic cells, and TNF-a-secreting cells, which could lead to a more severe inflammatory response135.
It is still unknown to what extent children transmit SARS-CoV-2. This understanding is of great importance for all countries to ensure safe reopening measures for schools. The sum of a few small epidemiological studies with children, with a few dozen cases, concludes that they are not the transmission source. Rather they might acquire the virus from adults in the great majority of cases136. However, larger pediatric cohorts are needed to determine the incidence of SARS-CoV-2 in children. For example, the Human Epidemiology and Response to SARS-CoV-2 from the NIH; the Pediatric Tuberculosis Network European Trials Group; the Kids Corona from Sant Joan de Déu (SJD) Barcelona Childrens Hospital, among others, where they are following kids and their families for several weeks, and in summer camps, to determine pediatric transmission115,125,137. Preliminary results of the home transmissibility study from the SJD Childrens Hospital, where 724 children living with a COVID-19 positive parent were monitored, showed that the percentage of infected children is very similar to adults. This serological epidemiological study suggests that children are infected at the same rate as adults when exposed to an infection source. However, the disease is milder in children than adults since over 99% of minors showed mild or no symptoms137. Therefore, it is possible that the reason for the earlier results from China, Italy, and the USA, which reported low rates of pediatric COVID-19 cases138-140, was testing mostly symptomatic cases. It is important not to dismiss without careful analysis the possibility of children, especially older children, being asymptomatic spreaders since school outbreaks have already been reported in some countries, such as New Zealand, Chile, and Israel141.
Recently, SARS-CoV-2 has also been implicated as the likely cause of a newly recognized pediatric syndrome. In late April, cases of rare symptoms similar to Kawasaki disease (KD) and toxic shock syndrome associated with COVID-19 were reported in children from the UK and Italy142,143. Shortly after, New York City also reported several cases, and the NYC Health Department and CDC alerted doctors of a multisystem inflammatory syndrome in children (MIS-C)144-146, which was called Pediatric Inflammatory Multisystem Syndrome temporally associated with SARS-CoV-2 (PIMS-TS) in Britain. Cases have now been reported in France, Switzerland, and Spain, and most affected children did not have underlying comorbidities147,148. Patients were positive for SARS-CoV-2 either by PCR or antibodies, but symptoms presented as a post-infection condition and not during the acute infection in most cases. Even when the epidemiologic evidence implicates SARS-CoV-2 as the most likely cause of MIS-C, causality has not yet been established. MIS-C symptoms resemble toxic shock syndrome, KD, and secondary hemophagocytic lymphohistiocytosis/macrophage activation syndrome145,149,150. This condition is characterized by prolonged fever (> 4 days) without a clear cause, inflammation symptoms such as rashes, conjunctivitis, swollen hands and feet, and swollen lymph nodes. The cytokine expression pattern observed in MIS-C suggests an IFN signaling component, along with IL-6 and IL-10 production, similar to KD and acute pulmonary COVID-19 infection. However, the lack of elevated TNF-a or IL-13 levels may differ from acute pulmonary COVID infections144. GI symptoms such as abdominal pain, vomiting, and diarrhea are common. Inflammation of blood vessels, including coronary arteries, has been observed, and cardiac problems tend to show earlier than in KD. Treatments used so far include steroids and intravenous immunoglobulin, high doses of aspirin, and antibiotics. The long-term effects are still unclear, but several patients tend to develop heart problems and low blood pressure, suggesting that children with serious heart damage will most probably need monitoring145. The increasing prevalence of MIS-C is suggestive of a delayed hyperimmune response to SARS-CoV-2 infection. However, the exact incidence of MIS-C after asymptomatic or mildly symptomatic infection with SARS-CoV-2 is not known.
Risk factors
In the earliest report from Wuhan, China, risk factors for mortality in adult patients showed that 48% of hospitalized patients had at least one comorbidity. Hypertension was the most common (30%), followed by diabetes (19%) and coronary heart disease (8%). However, they reported that in-hospital death is associated with older age, a higher Sequential Organ Failure Assessment score, and D-dimer levels > 1 mg/ml on admission151.
In the quest to further understand how viral and host factors relate to the clinical outcome of COVID-19 patients, a study analyzing clinical, molecular, and immunological data from 326 Shanghai positive patients was conducted, including an analysis of the isolated genomic sequences from the viral samples. They found that patients were exposed to different viral genetic variants in the early phases of the outbreak. However, this situation did not affect the patients outcome. The risk factors that they found to be predictive of disease progression were lymphocytopenia, especially the reduction of CD4+ and CD8+ cells in patients upon hospital admission, and an extensive depletion of CD3+ T lymphocytes, linked to spikes in cytokines such as IL-6 and IL-8, associated with an adverse outcome. A higher risk for disease progression was associated with co-existing conditions such as age, comorbidities, and gender, with a higher risk for males. In this analysis, advanced age and lymphocytopenia were the two major independent risk factors, showing that disease severity determinants were related to the host and not to the viral genetic variant that infected them152.
The most extensive risk assessment analysis so far has been performed in England, with the OpenSAFELY platform, where they included more than 17 million people (40% of all their patients) linked to almost eleven thousand COVID-19-related deaths. In this analysis, most deaths were associated with male gender, advanced age, poverty, diabetes, obesity, and severe asthma, among other respiratory, neurologic, and chronic conditions. Strikingly, age was shown to be one of the highest risk factors: people older than 80 years were shown to be 20 times more likely to die from COVID-19 than 50-year-old people and 100 times more likely than those under 40 years of age. Hypertension as a risk factor was strongly related to age, obesity, and diabetes, and its risk factor ratio diminished when adjusted to the last two. Hypertension was also of greater risk in people up to 70 years of age and lower risk at older ages. Importantly, an association or the risk of death was also observed with nonwhite race and ethnicities, especially affecting Black and South Asian. This observation was sustained even after the adjustment of other risk factors, showing that only a small part of the excess risk is explained by the higher prevalence of medical problems and poverty153.
The ongoing COVID-19 pandemic caused by SARS-CoV-2 has infected more than 15 million people, resulting in over 600 thousand deaths in 188 countries, as of July 23, 2020. The pandemic has challenged the public health systems in all countries, with profound economic and political consequences, to a much greater extent, but with many parallels with the pandemic that was previously caused by SARS-CoV154. Much has been learned by the scientific and medical community about the biology, epidemiology, and pathogenesis of SARS-CoV-2 in the last 7 months; however, the evidence accumulated through nine decades of research on CoVs has served as the foundation for this new knowledge. Much of what was previously known for animal CoVs and the previously identified HCoVs has been confirmed for SARS-CoV-223. Research on SARS-CoV-2 has generated valuable but few new insights into the basic molecular biology and host-cell interactions of CoV, as much detail had been obtained previously. However, as SARS-CoV-2 catapults CoVs into notoriety, many new findings are likely to emerge in the near future, including a deeper understanding of the mechanisms that drive genome recombination and make CoVs highly adaptable to changes in tissue and host tropism and zoonotic episodes. Understanding the viral mechanisms responsible for alterations of cell functions during the infection will require the characterization of viral proteins that induce remodeling of cellular membranes during the formation of CoVs ROs, as these compartments direct viral genome replication and expression, conceal viral macromolecules from defense mechanisms, and are likely to regulate the innate cellular response. Although the evidence suggests that the immune system can generate protection against SARS-CoV-2, the production of antibodies seems to be short-lived, and the key role of the cellular response should be studied in further detail. Many aspects of the immunological response need to be further explored, as the symptoms and outcomes from the disease vary widely. The ability of SARS-CoV-2 to infect and replicate in the upper respiratory tract may be associated with milder symptoms but is likely to result in more efficient transmission. More severe COVID-19 symptoms may result from the lower respiratory tract infection, where the virus can also replicate. Moreover, it is clear that SARS-CoV-2 can infect other organs and is responsible for several extrapulmonary symptoms, although both organ and endothelial damage may be caused by the viral infection or the exacerbated hyperinflammatory immune response. Reports of coagulopathies in severe cases have been increasing, where clotting may be triggered by the viral infection or the ensuing inflammation. Although evidence suggests that children are mostly asymptomatic or have mild disease, much remains to be determined in terms of the age-groups at higher risk and may play important roles in transmission. Fewer infections or diseases in children may be due to differences in the pediatric immune response or differences in the susceptibility of infection, which may be linked to lower levels of ACE2 compared to adults. It is important to consider that asymptomatic children have been underrepresented in epidemiological studies, and different symptoms or disease presentations may be present in children, such as the inflammatory pediatric syndrome (MIS-C) that has been increasingly reported. Symptoms of MIS-C are similar, not identical, to KD and toxic shock syndrome. Many risk factors have been associated with severe or lethal outcomes of COVID-19, including obesity, diabetes, male gender, older age, and blood pressure-related diseases. However, social and racial factors (the latter, for the most part, probably related to race-related social inequalities) are now also considered to be important.
Clearly, important lessons can be learned from the SARS-CoV-2 pandemic: viral pandemics represent a permanent threat to human health, and the increased exposure of human populations to wild species of animals, either through commerce or occupation of new ecological niches, will increase the chances of zoonotic events. In addition, the ever-increasing human populations and continued occupation of new habitats, and the increased human mobility will contribute to the emergence of zoonotic viruses that infect humans. However, the accumulated knowledge of the biology and evolution of viruses can be used to understand fundamental aspects of viral emergence so that predictions can be made for potential emerging viruses155. For decades, virologists knew well that CoVs had zoonotic pandemic potential. However, no advances in multiple vaccine candidates were accomplished further than animal testing because they were deemed unnecessary and perhaps commercially unattractive. Therefore, it will be of foremost importance to establish a continued dialogue between the scientific community, health authorities, and policymakers so that the knowledge that is gained through scientific research can be harnessed for social benefit by ensuring preparedness of health systems capabilities and availability of effective vaccines and antivirals.

