











 nueva página del texto (beta)
nueva página del texto (beta) Español (pdf)
Español (pdf)
 Artículo en XML
Artículo en XML Referencias del artículo
Referencias del artículo
 Enviar artículo por email
Enviar artículo por email Citado por SciELO
Citado por SciELO  Similares en
SciELO
Similares en
SciELO 
 Permalink
PermalinkIntroducción
Los soplos cardiacos (SC) se presentan hasta en el 77% de los recién nacidos (RN) y pueden ser la manifestación inicial de una cardiopatía congénita (CC). Sin embargo, distinguir entre un SC inocente y un SC patológico en la etapa neonatal es un reto1. La incidencia de las CC es de 8/1,000 nacidos vivos (nv), y cada 2/1,000 nv presentan CC críticas (CCC). Las CCC requieren cirugía o cateterismo cardiaco dentro de los primeros 28 días de vida.
En los EE.UU. y países desarrollados, el 50% de las CCC no se detectan al nacimiento; el porcentaje se estima aún mayor en México y otros países en desarrollo. Actualmente, representan el 50% de todas las muertes en RN con malformaciones congénitas2. El tronco arterioso común (TAC) es una CC cianógena que presenta un solo vaso arterial y da lugar a la aorta, arteria pulmonar y arterias coronarias, una válvula troncal común y una comunicación interventricular (CIV)3. El TAC tiene una incidencia de 7/100,000 nv, y representa el 4% de las CCC4. Sin una corrección quirúrgica temprana, más del 80% de los pacientes fallecen antes del primer año de edad5. Para la detección temprana del TAC y otras CCC, el tamizaje por oximetría de pulso en RN es eficaz, no invasivo y de bajo costo, y además es capaz de detectar del 50 al 70% de los casos2.
Caso clínico
Paciente de sexo femenino que nació a las 38 semanas de gestación por vía abdominal a causa de sufrimiento fetal agudo, secundario a doble circular del cordón umbilical. Al nacimiento, se detectó un SC y se diagnosticó como inocente, aunque no se realizaron estudios para su análisis ni oximetría de pulso. Después de 72 horas de vigilancia sin presentar complicaciones perinatales, la paciente egresó del hospital junto con su madre. En consultas posteriores de atención primaria no se corroboró dicho diagnóstico. Más adelante, a los 4 años de edad, al escalar una pendiente, la paciente presentó tos, cianosis perioral, disnea y fatiga, por lo que se solicitó atención médica. Los síntomas presentados, más la presencia de un SC y palpitaciones, fueron los motivos de referencia al tercer nivel. Al recibir atención en Cardiología Pediátrica, en la exploración física se detectó aumento del diámetro anteroposterior del tórax, precordio hiperdinámico e impulso paraesternal izquierdo. En la auscultación cardiaca se identificó un chasquido de apertura intenso en el borde esternal superior izquierdo, seguido de un soplo expulsivo grado III/VI con irradiación al cuello; segundo ruido único (IIp), seguido de un soplo diastólico grado III/VI6. En las extremidades, se observó cianosis ungueal (2/4) y pulsos amplios. El porcentaje de saturación de oxígeno por oximetría de pulso (SpO2) fue del 84%, peso de 17.5 kg y talla de 102 cm (P50 y P15, según la Organización Mundial de la Salud). Al interrogatorio dirigido, la madre refirió cianosis ungueal desde el nacimiento. En la radiografía de tórax se detectó cardiomegalia (índice cardiotorácico de 0.53), arco pulmonar prominente, arco aórtico derecho y flujo pulmonar incrementado (Fig. 1). El electrocardiograma registró una frecuencia cardiaca de 100 latidos/min, ritmo sinusal, eje cardiaco a 120°, crecimiento auricular y ventricular derecho.
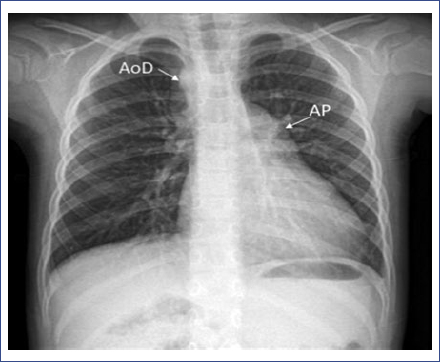
Figura 1 Radiografía de tórax posteroanterior. Se observa cardiomegalia (índice cardiotorácico de 0.53), arco pulmonar prominente (AP), arco aórtico derecho (AoD) y flujo pulmonar incrementado.
El cuadro clínico describió una CC cianógena con cardiomegalia con cortocircuito bidireccional, y sugirió la presencia de TAC. El ecocardiograma transtorácico (ETT) confirmó un TAC tipo I, según la clasificación de Collette y Edwards3 (Fig. 2), válvula troncal trivalva e insuficiencia valvular troncal (IVT) severa, CIV subpulmonar (8.35 mm) y arco aórtico derecho (Fig. 3). Se realizó una prueba de vasorreactividad pulmonar (PVP) por medio de cateterismo cardiaco derecho para evaluar la reversibilidad de las resistencias vasculares pulmonares (RVP). Los resultados obtenidos fueron iRVP de 3.56 UW/m2 (unidades Wood/m2) e iRVP/iRVS (índice de resistencia vascular pulmonar/ índice de resistencia vascular sistémica) de 0.33 (Tabla 1), concluyendo hipertensión arterial pulmonar (HAP) secundaria al hiperflujo pulmonar y vasorreactividad pulmonar. Finalmente, se decidió la corrección quirúrgica.
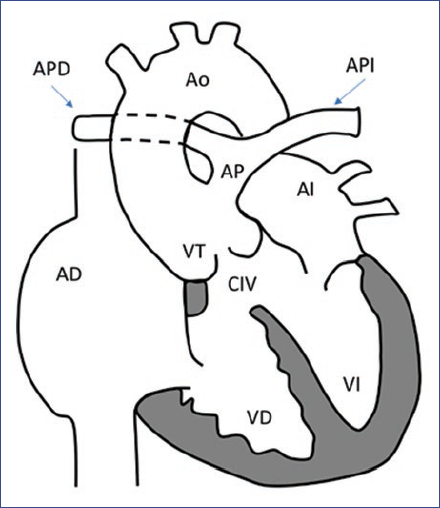
AD: aurícula derecha; AI: aurícula izquierda; Ao: aorta; AP: arteria pulmonar; APD: arteria pulmonar derecha; API: arteria pulmonar izquierda; CIV: comunicación interventricular; VD: ventrículo derecho; VI: ventrículo izquierdo; VT: válvula troncal.
Figura 2 Tronco arterioso común tipo I, según la clasificación de Collett y Edwards, la cual se basa en el origen de las arterias pulmonares. En el tipo I, la arteria pulmonar (AP) principal surge de la raíz troncal y se bifurca en arteria pulmonar derecha (APD) e izquierda (API); en el tipo II, la APD y la API surgen de un origen separado de la cara posterior de la raíz troncal; en el tipo III, cada arteria pulmonar surge independientemente de las caras laterales de la raíz troncal; en el tipo IV, no hay arterias pulmonares verdaderas presentes, y el flujo sanguíneo pulmonar es abastecido a través de vasos colaterales aortopulmonares. (Tipo II-tipo IV no se muestran en la figura).
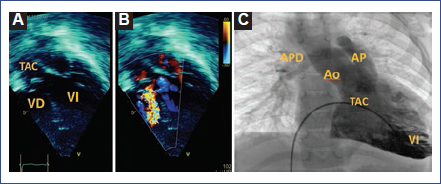
Figura 3 Ecocardiograma transtorácico bidimensional (cinco cámaras). A: Tronco arterioso común tipo 1. B: Ecografía Doppler a color que muestra insuficiencia severa de la válvula troncal. C: Ventriculografía izquierda del TAC que muestra la arteria pulmonar principal y aorta que confluyen en un tronco común. Además, se observa la tinción de las arterias pulmonares derecha e izquierda. Ao: aorta; AP: arteria pulmonar; APD: arteria pulmonar derecha; TAC: tronco arterioso común; VD: ventrículo derecho; VI: Ventrículo izquierdo.
Tabla 1 Valores hemodinámicos obtenidos por cateterismo cardiaco derecho
| FiO2 (21%) | FiO2 (100%) | |
|---|---|---|
| RIAP PAS (mmHg) PAM (mmHg) PAD (mmHg) |
115 64 42 |
97 67 41 |
| RDAP PAS (mmHg) PAM (mmHg) PAD (mmHg) |
95 64 42 |
96 67 45 |
| AoD PAS (mmHg) PAM (mmHg) PAD (mmHg) |
115 66 42 |
124 69 41 |
| UW iRPa (UW) iRVa (UW) Qp:Qs |
11.82 UW 9.72 UW 0.67:1.0 |
3.56 UW 10.54 UW 2.2:1.0 |
AoD: aorta descendente; FiO2: fracción inspirada de oxígeno; RPa: índice de resistencia pulmonar arteriolar; iRVa: índice de resistencia vascular arteriolar; PAD: presión arterial diastólica; PAM: presión arterial media; PAS: presión arterial sistólica; RDAP: rama derecha de la arteria pulmonar; RIAP: rama izquierda de la arteria pulmonar; iQp:Qs: relación de gasto pulmonar con gasto sistémico; UW: unidades Wood.
Procedimiento quirúrgico
Detrás del esternón y anterior al corazón se encontró tejido glandular correspondiente al timo. De la cara superior del ventrículo izquierdo emergía un tronco único, el cual originaba la aorta, que seguía un trayecto posterior y derecho; lateral e inferior al origen de la aorta surgía el tronco pulmonar (~ 15 mm) con trayecto anterior e izquierdo, dando lugar a las ramas pulmonares derecha e izquierda, respectivamente. Las arterias coronarias derecha e izquierda tenían origen en los senos posteriores del TAC, respectivamente. Por debajo de la válvula troncal (constituida por una valva anterior y dos posteriores), se localizaba una CIV infundibular (11 mm). No se encontraron defectos septales interauriculares. Se realizó timectomía subtotal, restitución de la continuidad del ventrículo derecho a la arteria pulmonar colocando xenoinjerto de 16 mm Contegra® (Medtronic, Minneapolis, MN, USA), plicatura subcomisural con resuspensión de las valvas sigmoideas troncales y cierre de la CIV con parche de poliéster. El tiempo de circulación extracorpórea fue de 169 min y el pinzamiento aórtico de 126 min. El ecocardiograma transesofágico posquirúrgico destacó insuficiencia valvular troncal leve.
Evolución postoperatoria y seguimiento
Durante el postoperatorio, la paciente requirió ventilación mecánica por 4 días. Además, presentó crisis de HAP, manejada con óxido nítrico inhalado (iNO) y citratro de sildenafilo (Revatio®, Viagra®, Pfizer Japan, Tokyo, Japan). Previo al egreso hospitalario, se evaluó la función cardiaca mediante ETT, en la que destacó IVT moderada.
En el seguimiento a un año, se mantuvo estrecha vigilancia de la función biventricular debido a IVT y HAP persistente; se manejó con inhibidor de la enzima convertidora de angiotensina (IECA), diuréticos y sildenafilo. Sin embargo, la paciente continuó con hiperactividad precordial, Ilp intenso, cianosis ungueal y SpO2 del 84% (Fig. 4). Las evaluaciones por ETT mostraron falla ventricular derecha e IVT moderada. Por consiguiente, se realizó una segunda PVP, la cual mostró HAP severa aún con vasorreactividad pulmonar. Por lo anterior, se agregó Bosentan® (Actelion, USA) y se reajustaron las dosis del tratamiento previo. Simultáneamente, se tomaron niveles plasmáticos de NT-proBNP (prohormona N-terminal del péptido natriurético tipo B), y se obtuvo un resultado de 822 pg/ml (valor normal ≤ 125 pg/ml). En un periodo de 18 meses el NT-proBNP se redujo a 60 pg/ml. Actualmente, la HAP está controlada y la paciente continúa en estrecha vigilancia de la función cardiaca, hepática y renal.

Discusión
El 95% de las CC se encuentran en población de bajo riesgo, por lo cual es necesario ofrecer a la población general un tamizaje cardiaco efectivo en edades tempranas7. Las CC se pueden diagnosticar mediante ecocardiografía fetal (ECF) entre la semana 18 y 24 de gestación, con lo que se detecta el 80% de los casos2,7. Actualmente, la ECF debe ser un estudio de rutina como parte de la evaluación del segundo trimestre en todas las pacientes embarazadas; la indicación para aquellas con factores de riesgo para CC es de ECF avanzada7.
En un estudio retrospectivo realizado por Cloete, et al., en Nueva Zelanda, se reportó una tasa de detección prenatal para CCC del 77% por ECF para el año 2015. Las CCC diagnosticadas por ECF fueron TAC, ventrículo izquierdo hipoplásico, tetralogía de Fallot y atresias valvulares, mientras que el diagnóstico por ECF para la transposición de grandes arterias y lesiones obstructivas izquierdas fue del 68% y del 53%, respectivamente. Además, la detección de CCC prenatal fue mayor en fetos con anomalías múltiples y menor en fetos con CCC aisladas2,8. En México, se desconocen las cifras de detección prenatal de CCC. Para el año 2017, según el reporte del Instituto Nacional de Estadística y Geografía (INEGI), el 50% de las muertes en menores de 1 año por malformaciones congénitas se debieron a CC, de las cuales, el 52% ocurrieron en el periodo neonatal debido a CCC9. Esto refleja que la mayoría de la población en México aún no tiene acceso al tamizaje prenatal ni posnatal para la detección de CCC.
Por lo tanto, el segundo momento para detectar una CC es al nacimiento. Alrededor del 77% de los RN pueden tener un SC; sin embargo, cerca de la mitad de los pacientes con CCC no lo presentan1. En el RN es difícil distinguir un SC inocente de uno patológico y la evaluación visual de cianosis es poco precisa2. El diagnóstico de CC a partir de un SC en el RN posee una sensibilidad del 44% para pediatras y del 80% para neonatólogos o cardiólogos pediátricos, con una especificidad del 33 al 90%1.
En el caso descrito, se diagnosticó un SC inocente durante las primeras 72 horas de vida de la paciente; sin embargo, el SC inocente se presenta sin fenómenos agregados y en ausencia de cianosis o insuficiencia cardiaca6. Desde el nacimiento, los pacientes con TAC presentan como primer ruido cardiaco un clic de eyección o chasquido de apertura (patognomónico de esta cardiopatía), junto a un soplo sistólico; el segundo ruido cardiaco es intenso, y cuando hay IVT, está asociada a un soplo diastólico. Además, cursan con cianosis ligera o moderada4, que puede ser difícil de reconocer, ya que es mayormente visible cuando la SpO2 ≤ 85%2.
EL ETT es el método no invasivo de elección en el diagnóstico de las CC; sin embargo, es una herramienta costosa y no es práctico para todos los RN. Por ello, desde 2011, el Comité Asesor de Enfermedades Hereditarias en Recién Nacidos y Niños, en colaboración con la Asociación Americana de Pediatría, la Asociación Americana del Corazón y la Fundación del Colegio Americano de Cardiología, propone el uso de la oximetría de pulso como tamizaje (POS, por sus siglas en inglés) para la detección de CCC en todos los RN después de las 24 horas de vida o lo más cercano al alta hospitalaria (esta medida fue implementada en los EE.UU. desde septiembre del 2011). En 2012, Thangaratinam, et al. realizaron un metaanálisis en el que se incluyeron 229,421 RN asintomáticos evaluados por POS. Se detectó del 50 al 70% de las CCC en RN después de las 24 horas de vida, con una especificidad del 99.8%, sensibilidad del 76.5%, falsos positivos del 0.05% después de las 24 horas de vida, y los falsos negativos fueron RN con coartación aórtica y estenosis aórtica severa. Se estima que la detección para transposición de grandes arterias por POS es del 100%2. Por lo anterior, si durante la hospitalización de la paciente (72 horas) se hubiera colocado el oxímetro de pulso, se habría evidenciado hipoxemia leve, y al aplicar el algoritmo para la detección de CCC más la presencia de un SC, se hubiera logrado un diagnóstico temprano10.
La cianosis que presentan los portadores de TAC se debe a la mezcla de sangre saturada y desaturada a nivel de la CIV, la cual sale del corazón a través del tronco único para distribuirse entre la aorta y las ramas pulmonares. La presión de perfusión es similar en la vasculatura sistémica y pulmonar, mientras que el volumen sanguíneo dependerá de la relación de las resistencias vasculares sistémicas y RVP. Más del 80% de los pacientes fallecen antes del año de edad, secundario a la asociación con anomalías que llevan a falla cardiaca, tales como arterias pulmonares de grueso calibre, arco aórtico interrumpido o hipoplásico, origen anómalo de las arterias coronarias, estenosis de la arteria pulmonar y conducto arterioso permeable4,11. El arco aórtico derecho puede acompañarse o no de lesiones obstructivas. Por otro lado, se ha asociado el TAC con la deleción del cromosoma 22q11.2 en el 75% de los casos4.
En este caso, la paciente no era portadora de la cromosomopatía, las arterias pulmonares eran de calibre delgado y el arco aórtico derecho no tenía obstrucciones; por lo tanto, estas características aumentaron su sobrevida. No obstante, la exposición prolongada a la sobrecarga de presión y volumen en la vasculatura pulmonar llevaron al incremento progresivo de RVP4,12.
Después del primer año de edad, la mortalidad en pacientes con TAC se asocia con el desarrollo precoz e irreversible de HAP5,4,11. Para definir la posibilidad de corregir una CC con HAP por cirugía, se realiza cateterismo cardiaco derecho para obtener iRVP y la relación iRVP/iRVS. Un valor de iRVP < 6 UW/m2 e iRVP/ iRVS < 0.3 son indicativos de cirugía y de buen pronóstico posquirúgico12.
Los resultados del cateterismo cardiaco derecho en la paciente fueron iRVP = 11.82 UW/m2 e iRVP/iRVS = 1.21, ambos por encima del valor aceptable, por lo que se realizó la PVP y se observó una disminución del 70% en ambos valores (iRVP = 3.56 UW/m2 y iRVP/iRVS = 0.33). La disminución del 20% en la iRVP y en el iRVP/iRVS después de la PVP representan todavía un buen pronóstico posquirúrgico12. En la mayoría de los pacientes con TAC, como este caso, la variante anatómica tipo 1 (TA con arterias pulmonares confluentes)3 y la válvula troncal trivalva son las características más comunes. La IVT severa, que es rara (< 5%), es indicativa de corrección quirúrgica temprana, ya que es el principal riesgo de mortalidad preoperatoria. La técnica quirúrgica más utilizada es la de Rastelli (corrección biventricular) con interposición de un tubo valvado, que tiene baja mortalidad aun en etapas tardías del TAC. Se estima que la sobrevida posterior a la cirugía es del 87.8% a 5 años. Hasta el 92.9% de estos pacientes son libres de una reintervención quirúrgica por 5 años. La complicación postoperatoria tardía más frecuente es la estenosis del tubo valvado. Las técnicas de reparación de la válvula troncal, como la suspensión comisural, son efectivas y duraderas. En el caso de IVT leve a moderada, se brinda tratamiento médico para evitar falla ventricular izquierda. Sin embargo, si la IVT empeora, el reemplazo valvular es inevitable, y presenta una mortalidad del 66% en reemplazo valvular y del 25% al colocar un homoinjerto4,5.
Una vez establecida la HAP, la corrección quirúrgica de CC presenta alto riesgo de crisis de HAP en el postoperatorio inmediato, pero responde favorablemente al iNO y sildenafilo vía oral13. No obstante, la [endotelina-1]p (vasoconstrictor endotelial) continúa elevada, llevando a HAP persistente. Ante esto, algunos pacientes se conservan estables por un periodo de 3-5 años; en otros, el deterioro es rápido, lo que conduce a la falla ventricular derecha y muerte14.
El manejo en estos pacientes es complejo. Actualmente, las recomendaciones se basan en estudios de adultos y en consenso de expertos15. El uso de diuréticos, oxígeno, anticoagulación, digoxina o IECA debe considerarse de forma individual. Ante el deterioro rápido de HAP, se requiere repetir la PVP con iNO. La respuesta negativa a la PVP dirige al uso de bosentán (antagonista dual del receptor endotelina A y B) como tratamiento de elección o sildenafilo (inhibidor de la fosfodiesterasa 5), considerando también la terapia combinada o secuencial, especialmente en aquellos pacientes que se deterioran con monoterapia15. Además, el uso de bosentán a largo plazo (> 6 meses) ha demostrado disminuir la RVP y controlar la remodelación vascular14.
El BNP, NTproBNP y el ácido úrico son los biomarcadores recomendados para la estratificación de riesgo en HAP15,16. La sobrecarga de volumen ventricular derecho es el principal disparador de la producción de BNP. Los valores de BNP ≥ 104 pg/ml están asociados con un mayor riesgo de muerte, y aquellos < 50 pg/ml, con una mejor supervivencia17. El NT-proBNP es el mejor predictor de mortalidad en HAP, ya que se correlaciona directamente con la RVP y la presión arterial pulmonar media, además de que ha demostrado ser un marcador de eficacia para el tratamiento de HAP. Los pacientes con valores de NT-proBNP ≥ 1,256 pg/ml al diagnóstico tienen un mal pronóstico16. El ácido úrico también es un predictor de pronóstico en HAP, por lo que se recomienda su monitorización en el seguimiento de estos pacientes15.

