











 text new page (beta)
text new page (beta) English (pdf)
English (pdf)
 Article in xml format
Article in xml format Article references
Article references
 Send this article by e-mail
Send this article by e-mail Cited by SciELO
Cited by SciELO  Similars in
SciELO
Similars in
SciELO 
 Permalink
PermalinkIntroduction
Autosomal recessive osteopetrosis Type I (ARO1, OMIM 259700) is a rare multisystem bone disease, being the most severe phenotype of the various osteoclast disorders. In this disease, insufficient calcified cartilage resorption interferes with the normal turnover of mature bone. Affected individuals usually die in the first 10 years of life1. The cause of death usually is related to bone marrow failure. To date, seven genes at least have independently been linked in the etiology of autosomal recessive osteopetrosis, among which TCIRG1, CLCN7, and OSTM1 have been reported as the most frequently affected. Incidence is estimated to be 1 in 100,000 to 1 in 500,0002.
ARO1 is associated with premature infantile death. In newborns, it can be diagnosed in an early fashion (as early as 10 days of age). The disease hallmarks are mainly pathologic fractures, bone marrow failure, and cranial nerve compression due to impaired bone turnover metabolism and inability to widen cranial nerve foramina.
Case report
An 8-month-old infant was received for evaluation. Perinatal history relevant for being born to a fourth pregnancy of non-consanguineous parents (a three-generation pedigree was conducted to rule out consanguinity) and obtained through vaginal delivery at 38 weeks of gestational age. Bodily measurements revealed weight (4,200 g, fifth percentile) and height (72 cm, seventh percentile).
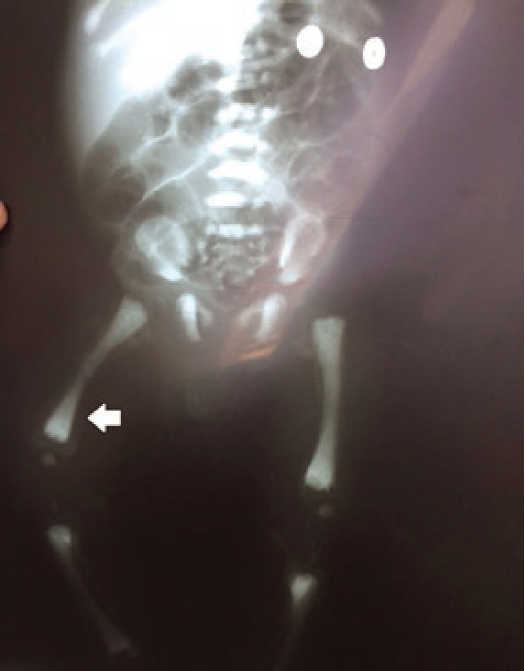
The mother noted an absence of gaze fixation, pallor, petechiae, poor feeding skills, and failure to grow. He was initially diagnosed as hypothyroid and given thyroxine for 6 months with no apparent benefit. A complete blood count was obtained and cytopenias (leukopenia without neutropenia, anemia, and thrombocytopenia). On examination, the infant was found with hypotonia, abnormal eye movements, depressed nasal bridge, and hepatomegaly without splenomegaly. An abdominal ultrasound measured a normal spleen and an enlarged liver with 345 cc estimated volume. A hematology consultation was sought. On peripheral smear, abnormal azurophilic granules in lymphocytes and neutrophils were apparent (Fig. 1). The initial bone marrow evaluation resulted in a dry tap. On marrow blood, no blasts, Gaucher cells, or Niemann-Pick cells were identified. Due to lack of a definitive diagnosis, the patient was discharged and put on surveillance. Three months later, he was admitted on the basis of pancytopenia and a community-acquired pneumonia. In the second hospitalization, a new bone marrow aspiration with bone marrow biopsy was attempted an adequate sample with Trucut technique but could not be obtained due to the small size of the patient pelvis. A diagnosis of osteopetrosis was suggested and plain films were ordered. Marked opacity of bone in thorax and ribs, skull, and long bones was noted (Figs. 2 and 3). A “bone within bone” image was evident. Extended bloodwork revealed increased parathormone levels (85 pg/ml), normal calcium, and reduced phosphorus levels. Ancillary consultations were sought. Maxillary surgery reported maxillary and mandibular hypoplasia, with the absence of tooth eruption. Pediatric ophthalmology noted an absence of fixed gaze, horizontal nystagmus, and optic nerve pallor, suggesting bilateral optic nerve hypoplasia. A genetic consultation was sought for molecular confirmation of diagnosis due to inadequate bone marrow sample. With an informed consent, DNA was obtained from the proband and his parents, from an oral brush technique for the patient and in peripheral blood for parents. DNA was extracted through routine protocol and sent abroad for molecular study.

Figure 1 Peripheral blood smear. Anomalous lymphocyte inclusions. Optic microscopy. May-Grünwald-Giemsa (×100).

The sample was subjected to direct sequencing of the PCR amplified exons. Two pathogenic variants of the TCIRG1 gene were detected: on the maternal allele, a single nucleotide change (c.504-6 C>A), and on the paternal allele, a nine nucleotide deletion (c.1809_1818del), confirming a compound heterozygote status for the proband and the carrier state of both the parents.
The pathogenic variant on the maternal allele has previously been reported and is described as a change in the acceptor site of splicing machinery in exon 6, which produces abnormal splicing with frameshift effect.
Deletion on paternal allele has not been reported previously.
Bone marrow transplantation in a reference hospital was consulted for the case evaluation. They considered an intact visual function as a requirement for transplantation, as measured by visual-evoked potentials. The procedure was nonetheless offered to the patient, which declined to consent. At the time of manuscript writing, the patient was still alive but frequently afflicted by infections, cytopenias, and transfusion dependence.
Discussion
Malignant infantile osteopetrosis (MIO)
Classical clinical presentation of patients is described in the present case: generalized osteosclerosis, cytopenias, lymphadenopathy, or hepatosplenomegaly. Cranial nerve compression (anosmia, deafness, and blindness), lacks teeth eruption. Other modes of presentation are hydrocephalus3 or abnormal calcium and phosphorus metabolism4.
The diagnosis of this entity requires a high suspicion awareness. In this case, diagnosis was conducted in a late fashion (five months between first contact with the patient and the molecular analysis of TCIRG1). Diagnostic workup required anesthetic procedures for biopsies, bone marrow aspirations, and catheter placements. In this setting, finding a difficult airway is the norm and is attributable to cranial and mandibular abnormalities associated with the phenotype5.
Basic biology of osteoclasts
Osteoclasts are cells that specialize in the resorption process of mineralized matrix (for instance, bone, dentine, and enamel). It is possible that there are specific osteoclast populations adjusted to degradation of specific organic matrix types or associated with specific bones (for instance, cranium-specific osteoclasts vs. long bone osteoclasts).
These cells adhere to organic matrices and form a sealed zone between the membrane and the mineralized matrix. The vacuolar proton pump (VPP-ATPase) is necessary for proton pumping and acidification of the cell-matrix interphase. The matrix thus acidified experiments dissolution of hydroxiapatite and subsequent exposure of the organic component (such as Type I collagen). Subsequently, there is exocytosis of enzymes such as cathepsin K, which results in effective digestion of bone6.
The final result is the dissolution of bone and matrices to allow for remodeling in processes of growth, mobilization of calcium and phosphorus reservoirs, and tooth eruption, among others7.
The differentiation and maturation or osteoclasts require soluble mediators, but also cellular ligands. Two molecules are enough for inducing the transformation into osteoclast: macrophage colony stimulating factor and ligand of receptor for the activation of nuclear factor-kB.
Osteoclasts arise from cells belonging to the monocyte and macrophage lineage and then experiments cellular fusion which in turn produces a multinucleated cell.
TCIRG1 and the genetics of osteopetrosis
ARO1 is a genetically heterogeneous disorder, being described as an autosomal dominant, autosomal recessive, and X-linked.
TCIRG1 (T-Cell immune regulator 1, Gen ID: 10312): This gene codes for the A3 subunit of the VPP1, which is expressed on the ruffled border of the osteoclast membrane. It is localized to chromosome 11q13.2. Alterations on TCIRG1 represent approximately half the cases of ARO1. A3 subunit is composed of two functional domains: the hydrosoluble V1 and the membrane-spanning VO which performs ATP hydrolysis, activating the proton pump with subsequent acidification of the extracellular membrane. The A3 isoform is highly expressed on the osteoclast membrane8.
It plays a crucial role in bone development and remodeling. It is known of two transcriptional variants: OCC116 (NP_006010), which consists of 20 exons, but translation starts on exon 2, and TIRC7 (NP_006044), which is a splice product of exons 5 through 20 but with retention of intron 5. OCC116 is expressed mainly in osteoclasts, while TIRC7 is expressed in other tissues (where it might have a role in receptor recycling processes, e.g., insulin or transferrin receptors), and it is believed to play an essential role in T-cell development. ATP-dependent acidification is necessary for intracellular processes such as sorting of secreted proteins, zymogen activation, and receptor-mediated endocytosis.
Various types of TCIRG1 pathogenic variants have been described9: nonsense, insertion/deletion, frameshift, as well as defects of splicing sites throughout the whole gene10. Mutant homozygotes and compound heterozygotes have also been described11,12.
Other genes might experience pathogenic variants and lead to osteopetrotic phenotypes, such as CLCN7 (involved in chloride ion pumping), carbonic anhydrase type II, OSTM1, and PLEKHM1 (the latter being involved in the transport of acidified endosomal vesicles).
Treatment
Hematopoietic stem cell transplantation represents the definitive treatment of autosomal recessive MIO. Transplantation is curative because it replaces the patient’s hematopoietic system, and the osteoclast population is bone-marrow derived, differentiating from monocytic or occasionally dendritic cell precursors13,14.
It is preferred to conduct the procedure on an early fashion, preferably before the advent of cranial nerve compression (blindness, auditory, and strabismus), as this is generally nonreversible. Nonetheless, the presence of ophthalmic and auditory involvement should not contraindicate the procedure. Various case series have documented an improvement on growth (reinstallation of growth and resumption of endochondral and intramembranous ossification processes and tooth eruption15), hematopoiesis (remodeling of medullary cavity and resorption of bone and cavity cartilage), and resolution of hypocalcemia and hypophosphatemia (when bony calcium and phosphorus sinks are mobilized again) (Table 1).
Table 1 Therapeutic effects of hematopoietic stem cell transplantation
| Reduction of hepatosplenic volume |
| Appearance of osteoclast and trilinear hematopoietic differentiation |
| Resumption of tooth eruption |
| Resolution of rachitic bone changes |
Many conditioning regimens have been employed in transplantation for MIO: busulfan/cyclophosphamide, busulfan/cyclophosphamide/thiotepa, fludarabine/busulfan, antithymocyte globulin, as well as multiplicity of cell sources: bone marrow, peripheral blood, and cord blood. Regarding donor type, the procedure has been carried out on related HLA identical and alternative donors (non-related, identical, ande haploidentical) (Table 2).
Table 2 Selected topics regarding transplantation for osteopetrosis
| Most frequent donor type was HLA identical related |
| Median age at transplantation is 12 months |
| Most frequently used conditioning regimen was busulfan/cyclophosphamide |
| No difference in outcome with HLA identical versus alternative donors |
| Death rate of 50%, with main cause of death being infection related |
| Median age at diagnosis in the survivor population: 7 years |
| 70% of patients with some degree of cranial nerve compression |
| 10% defect in hearing or language development |
| Higher rate of vaso-occlusive disease and interstitial pneumonitis than for other hematopoietic stem cell transplantation indications |
| Higher clinically significant graft versus host disease (GII-IV) in alternative donors versus HLA identical |
| 65% of long-term survivors with acceptable functional outcomes |
HLA: human leukocyte antigen system
A recurrent immediate complication unique to the disease type is the appearance of hypercalcemia, due to bone mobilization. The calcium levels postransplantation peaked on days +9 to +29 throughout various reports. The frequency of this complication is about 35%. Treatment has required hyperhydration, steroids, and on occasion, bisphosphonates16.
Although transplantation for osteopetrosis is known to be performed in transplantation centers in Mexico, only one publication was found, in which the patient was transplanted with a reduced intensity approach17.
Patients considered poor candidates for hematopoietic stem cell transplantation can benefit from the use of interferon γ1B, which has demonstrated therapeutic effects in various series: rise in hemoglobin, reduction in number of infections, rise in trabecular bone, and bone marrow cavity partial restoration18.
Autosomal recessive osteopetrosis type I is an infrequent autosomal recessive entity. The diagnosis requires a high level of suspicion. The latter is essential, as an early diagnosis markedly improves access to treatment with hematopoietic stem cell transplantation in better overall condition. A previously non-reported deletion of TCIRG1 is described. General aspects of diagnosis, biology of the osteoclast population, and treatment of the disease are reviewed. The scarcity of publications of the procedure in Mexico is notable, though it is known that transplantation is carried away more frequently than it is reported.

