











 nueva página del texto (beta)
nueva página del texto (beta) Español (pdf)
Español (pdf)
 Artículo en XML
Artículo en XML Referencias del artículo
Referencias del artículo
 Enviar artículo por email
Enviar artículo por email Citado por SciELO
Citado por SciELO  Similares en
SciELO
Similares en
SciELO 
 Permalink
Permalink1. Introducción
Los tumores malignos de la vaina del nervio periférico (MPNST) son sarcomas de la vaina nerviosa derivados de las células de Schwann o células pluripotenciales de la cresta neural1, conocidos también como neurilemomas, schwannomas malignos, neurofibrosarcomas y sarcomas neurogénicos2. Los MPNST son tumores muy poco frecuentes, con una incidencia del 0.001% en la población general y del 0.16% en pacientes con neurofibromatosis tipo 1 (NF1)3. Representan aproximadamente el 10% de los sarcomas de tejidos blandos y, a pesar de su rareza, son de los tumores no rabdomiosarcomatosos más frecuentes en la edad pediátrica4, altamente agresivos y asociados generalmente con NF1 y con tratamiento previo con radioterapia5.
El objetivo de este trabajo fue presentar el caso de un MPNST retroperitoneal en una paciente sin neurofibromatosis, hacer una revisión de la literatura y discutir las posibilidades terapéuticas complementarias después del abordaje quirúrgico.
2. Caso clínico
Paciente de sexo femenino de 2 años y 10 meses de edad procedente de una zona urbana. Consultó por masa abdominal palpada por la madre 7 meses antes, con crecimiento rápido en los últimos 2 meses. No presentaba dolor, fiebre, pérdida de peso u otros síntomas asociados. Tenía el antecedente de estreñimiento crónico de larga data, sin antecedentes en la familia de neurofibromatosis ni de cáncer. Durante la exploración física se palpó una masa en el cuadrante inferior izquierdo del abdomen, con un tamaño de 15 x 10 cm, de consistencia dura, adherida a planos profundos y no dolorosa. No presentaba “manchas café con leche” ni lesiones que pudieran sugerir neurofibromas. El hemograma, deshidrogenasa láctica, ácido úrico, electrolitos séricos, la función renal y hepática fueron normales. La alfa fetoproteína y gonadotropina coriónica humana subunidad beta fueron normales, por lo que se descartó un tumor germinal.
La ecografía del abdomen total reportó una importante masa macrolobulada en el retroperitoneo, heterogénea, predominantemente hipoecoica, de 90 x 64 x 78 mm en sentido lateral, antero-posterior y transverso; no se identificaron crecimientos ganglionares retroperitoneales.
La tomografía axial computarizada (TAC) de abdomen contrastada mostró una masa heterogénea en la fosa iliaca izquierda, adyacente al foramen de conjunción L5-S1 ipsilateral. La radiografía de tórax fue normal.
Se realizó laparotomía y se tomó una biopsia por incisión. El aspecto macroscópico del tumor era sugestivo de fibrosarcoma, pero la muestra fue insuficiente para el diagnóstico, por lo que se tomó una nueva biopsia. El cirujano describió una masa retroperitoneal dura, de 15 x 10 cm, con gran componente vascular, bien delimitada, que emergía de planos posteriores aledaños a las vértebras lumbares y rechazaba hacia adelante vasos iliacos y órganos pélvicos. La cavidad abdominal estaba libre de tumor y con escaso líquido cetrino.
En la histopatología se observó neoplasia fusocelular atípica de difícil clasificación, sin evidencia de necrosis y con un conteo mitótico de hasta tres mitosis por 10 campos de gran aumento. No fue posible definir con certeza su histogénesis.
La inmunohistoquímica, realizada en el departamento de patología del Hospital universitario fundación Santa Fe de Bogotá (FSFB), mostró reactividad intensa y difusa en las células tumorales con los marcadores de proteína S100, CD34, y con los marcadores PGP 9.5 y sinaptofisina, positividad focal ocasional. El índice de proliferación celular (Ki67) fue del 5%. Los marcadores GFAP, HMB45, EMA, CK7, AE1/AE3, TLE1, ALK, desmina, CD117, CD99 y DOG 1 fueron negativos.
Las muestras fueron revisadas por el Departamento de Patología del Brigham and Women's Hospital (BWH) en Boston, Estados Unidos, que reportó “neoplasia de células fusiformes, moderadamente celular, en la cual las células tumorales tienen un citoplasma eosinofílico pálido y núcleos ligeramente ovoides o más ahusados. Presencia de mitosis dispersas multifocales. No se presentan pleomorfismo o necrosis significativas. Las inmunotinciones muestran positividad multifocal para la proteína S-100, positividad focal débil para SMA y células multifocalmente dispersas positivas para MDM2, mientras que GFAP es negativa. Los hallazgos morfológicos y de inmunofenotipo hacen pensar en un tumor maligno de la vaina del nervio periférico que parece ser de bajo grado”.
La paciente no cumplió con los criterios clínicos ni tenía historia familiar de NF1; el examen oftalmológico fue normal y Genética Clínica no consideró necesario tomar estudios genéticos especiales.
Se realizó la resonancia magnética nuclear (RMN) de abdomen contrastada previa a la resección tumoral, que mostró que la masa envolvía la arteria y vena iliaca izquierdas (Fig. 1).

Figura 1 Resonancia magnética de abdomen con contraste que muestra masa sólida de 107×80×76mm de diámetros craneocaudal, anteroposterior y transverso que ocupa hipogastrio, fosa iliaca y flanco izquierdos y gran parte del mesogastrio, pediculada desde los agujeros de conjunción L4-L5, L5-S1 y S1 izquierdos. Sólida, heterogénea, con necrosis, vascularizada, que envuelve parcialmente la bifurcación aórtica y totalmente la arteria iliaca primitiva izquierda y sus ramificaciones, al igual que la vena iliaca.
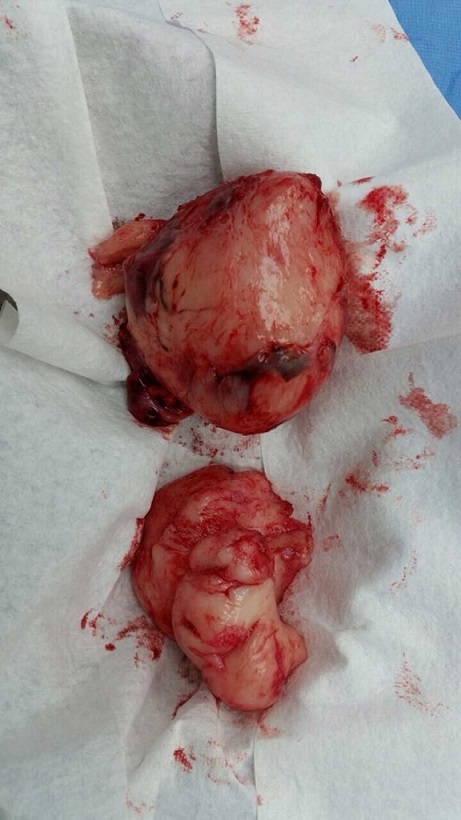
La paciente fue llevada nuevamente a cirugía para resecar el tumor. La masa rodeaba la arteria iliaca, el uréter, la arteria gonadal izquierda y la vena femoral; se extrajo aproximadamente el 80% del tumor (Fig. 2). Durante la cirugía presentó hemorragia y choque hipovolémico, por lo que se aplicaron transfusiones de glóbulos rojos empaquetados y vasopresores. La paciente fue internada en la Unidad de Cuidados Intensivos Pediátricos, de donde egresó 5 días después. La histopatología fue igual a la descrita anteriormente, pero con índice de proliferación del 15% medido con Ki67.
En el postoperatorio presentó dolor y edema del muslo izquierdo, con debilidad muscular, limitación para la aducción, disestesias en el dorso del pie izquierdo y pie equino ipsilateral que impedían la marcha; se descartó compromiso vascular por estudio Doppler. Se encontró compromiso nervioso a nivel de S1, además de desacondicionamiento físico; se inició terapia física y colocación de férula de posicionamiento. Se realizó una electromiografía que reportó lesión radicular L5-S1 vs. lesión del nervio ciático.
Actualmente, la paciente se ha recuperado de manera importante. Presenta cojera leve, pie caído, hipotrofia y paresia del miembro afectado. Se encuentra en seguimiento por fisiatría.
Durante los controles por oncología, los exámenes de laboratorio han sido normales. La RMN de abdomen tomada en el postoperatorio reportó una masa residual de 42.2 x 53.6 mm (Fig. 3). Tres meses después una ecografía mostró un aparente aumento del tamaño tumoral, de 73 x 54 x 58 mm. Posteriormente fue discutida en la junta de oncología pediátrica, y se decidió iniciar quimioterapia con ciclos de ifosfamida más doxorubicina, alternados con ifosfamida más etopósido.
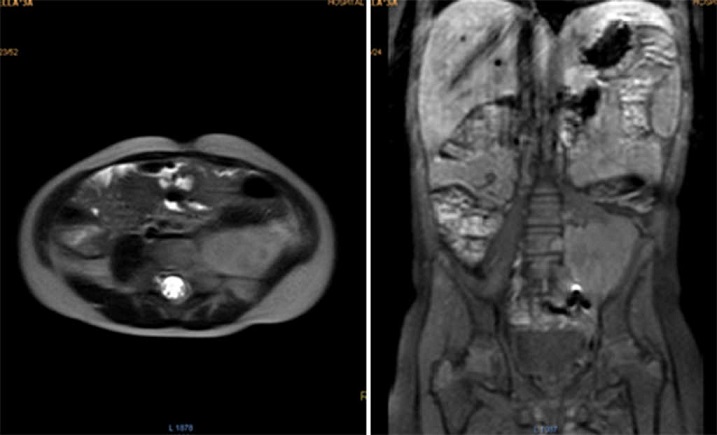
Figura 3 Resonancia magnética de abdomen con contraste mostrando a nivel paravertebral izquierdo masa de bordes definidos de aspecto heterogéneo en todas las secuencias, que mide aproximadamente 42.2×53.6mm, con ávido realce de la lesión con el medio de contraste.
De acuerdo con su evolución y respuesta, se definirá la posibilidad de hacer una segunda cirugía y complementar con radioterapia localizada. Se solicitó a los padres su consentimiento para la publicación del caso, guardando la debida confidencialidad.
3. Discusión
Los MPNST son tumores raros que generalmente ocurren entre los 20 y 50 años de edad, por lo que los casos publicados en niños son muy escasos4,5,6. La paciente reportada presentó una masa abdominal palpable desde los 27 meses de edad, con diagnóstico a los 34 meses, edad a la cual esta enfermedad es extremadamente rara. Bates y colaboradores7 encontraron un total de 139 pacientes menores de 19 años de edad con MPNST entre 1973 y 2009, y solo 5 casos en niños entre 1 y 4 años (3.6%), utilizando la base de datos del SEER (Survival Epidemiology and End Results). Este grupo de niños presentó una menor incidencia (0.19 por millón de personas-año, IC 95% 0.09-0.36) con una diferencia estadísticamente significativa respecto a los de 10 a 19 años (de 10-14 años con incidencia de 0.72, IC 95%: 0.52-0.97 y de 15-19 años con incidencia de 0.99, IC 95%: 0.75-1.28, p < 0.001). En el estudio no se encontraron diferencias significativas en los diferentes grupos étnicos, pero no se mencionaron datos específicos sobre hispanos. Otro estudio, que utilizó también la base de datos del SEER, comparó niños y adultos con MPNST de 1973 a 20081. En este estudio se menciona que el porcentaje de adultos blancos no hispanos fue el 68.3%, mientras que en los niños hubo una mayor proporción de negros e hispanos respecto a los blancos no hispanos (18.8%, 26.6% y 49.2%, respectivamente) con una diferencia estadísticamente significativa (p < 0.0001).
Desde el punto de vista de la presentación clínica, los datos reportados en la literatura incluyen un crecimiento tumoral lento, los pacientes son asintomáticos durante mucho tiempo y los signos de la enfermedad y las alteraciones sensitivo-motoras aparecen generalmente en una etapa avanzada8,9. En esta paciente, la enfermedad se manifestó como una masa asintomática de crecimiento lento sin hallazgos neurológicos.
Las imágenes diagnósticas más precisas son la TAC y la RMN, por encima de la ultrasonografía. La TAC es útil para evaluar la extensión del tumor y sus metástasis, pero la RMN es la prueba de elección porque puede revelar el nervio de origen y su relación con las estructuras vasculares, musculares y planos grasos8,10,11. La TAC realizada a la paciente mostró estrecha relación con los forámenes de conjunción L5-S1, lo cual se confirmó posteriormente con la RMN. Esto es consistente con que la mayoría de MPNST se originan de troncos nerviosos mayores, incluyendo el ciático, plexo braquial y sacro, por lo que las localizaciones más comunes son el tronco y las zonas proximales de las extremidades12,13. Una imagen de gran utilidad es la tomografía con emisión de positrones (PET Scan), la cual permite visualizar el metabolismo de la glucosa por las células tumorales, y es principalmente útil en demostrar la trasformación maligna de los neurofibromas plexiformes9.
El tamaño del tumor, de 100 x 150 mm, fue superior a la mediana reportada en niños con MPNST en tórax y abdomen en los Estados Unidos, y estuvo cerca de los mayores tamaños reportados en ese país (mediana de 85 mm con desviación estándar de 67 mm)1. En ese estudio se observó que había mayor mortalidad en las niñas, probablemente porque presentaban un mayor tamaño tumoral (mediana de 85 mm) comparado con el tamaño que presentaban los niños (mediana de 63 mm). El 57% de las mujeres tuvieron tamaños de, al menos, 70 mm, lo que solamente ocurrió en el 26% de los hombres; la causa de esta diferencia es desconocida. En la paciente reportada, la masa descrita rodeaba la arteria iliaca, el uréter, la arteria gonadal y la vena femoral izquierdas, impidiendo la resección total. La ecografía de control posterior sugiere un crecimiento tumoral importante (73 mm). Los MPNST más grandes pueden ser más difíciles de resecar completamente, lo cual se refleja en la mayor mortalidad de pacientes pediátricos con tumor grado III/IV en el tronco (HR: 12.53, IC 95%: 3.11-50.47) o clasificados como tumores no localizados por tener enfermedad regional o distante (HR: 0.53, p <0.001)1. Reportes de casos de diversos países y regiones podrían ayudar a determinar si estos tumores son realmente más grandes en las niñas en comparación con los niños.
Desde el punto de vista macroscópico, los MPNST son tumores voluminosos (> 10 cm) de localización profunda. Al corte semejan el aspecto de carne de pescado con focos de necrosis y hemorragia. Microscópicamente se ven lesiones altamente celulares dispuestas en fascículos, que pueden confundirse con fibrosarcomas o schwannomas hipercelulares, alternados con zonas mixoides hipocelulares que pueden parecer lesiones benignas14. Las células son fusiformes (similares a las células de Schwann) o pueden ser redondas (semejantes morfológicamente a fibrosarcomas o sarcomas de células redondas), con citoplasma poco prominente, núcleos hipercrómicos con nucléolos bien aparentes,9,14 mitosis frecuentes y necrosis focal15. Los reportes de patología de la FSFB y el BWH describen células fusiformes, con citoplasma eosinófilo pálido, núcleos atípicamente ovoides y mitosis multifocales dispersas.
En la inmunohistoquímica, la proteína S100 se ha identificado en aproximadamente el 50-90% de los MPNST, usualmente de forma focal2,6,10,15,16,17; es el marcador más utilizado para documentar diferenciación de la vaina del nervio periférico, pero también está presente en sarcomas sinoviales, schwannoma celular y melanoma fusiforme. CD34 es una proteína que está presente en algunos MPNST. CD56 y la proteína producto del gen 9.5 (PGP 9.5) son considerados antígenos sensibles para MPNST2. En el caso reportado se muestra una positividad multifocal para S100, y positividad focal para PGP 9.5 y sinaptofisina. EMA y CK7 fueron negativas; estas dos proteínas juntas tienen alta especificidad para sarcoma sinovial2. AE1/AE3, CD99, TLE1, frecuentes en ese sarcoma, también fueron negativos. El tumor presentó positividad focal débil para SMA, a diferencia de los leiomiosarcomas que tiñen fuertemente para ese antígeno. La desmina, que está presente en muchos leiomiosarcomas, fue negativa en este caso16.
En un estudio de factores de riesgo, investigadores chinos encontraron que cuando la proteína S-100 era negativa o débilmente positiva sus pacientes tenían mayor frecuencia de recurrencia y peor sobrevida, con diferencia estadísticamente significativa al compararlos con pacientes con S-100 positiva17.
La totalidad de las publicaciones acerca del tratamiento coinciden en que el objetivo fundamental del mismo es la extirpación quirúrgica del tumor, idealmente en forma completa, con márgenes quirúrgicas negativas, lo cual repercute en una mejor sobrevida libre de enfermedad (SLE), menor tasa de recurrencia y mayor sobrevida total (ST)4,12,13,14,18,19,20. Aunque ha habido gran controversia en cuanto a la utilización de la quimioterapia en el tratamiento de los MPNST, hay estudios que confirman su beneficio, especialmente con la combinación de doxorubicina e ifosfamida que resultó superior en diferentes esquemas de tratamiento, mejorando significativamente la SLE y ST en pacientes con tumores de alto riesgo21,22. Dada la limitación que tienen estos trabajos por ser retrospectivos, los autores sugieren la realización de estudios prospectivos aleatorizados para tener una mayor claridad en cuanto a la utilización de agentes quimioterapéuticos. Previamente, el grupo italo-alemán había confirmado el beneficio de la quimioterapia, especialmente de protocolos con ifosfamida, y en su trabajo recomiendan quimioterapia neo-adyuvante para pacientes con enfermedad imposible de resecar al momento del diagnóstico, con el fin de reducir el tamaño tumoral para realizar la cirugía y continuar el tratamiento con quimioterapia y radioterapia4. De igual manera, la utilización de la radioterapia como tratamiento adyuvante es controvertida; sin embargo, diferentes estudios demuestran un mayor control local con la utilización de la radioterapia, especialmente cuando hay enfermedad microscópica, aunque su beneficio cuando hay enfermedad macroscópica postquirúrgica es incierto4,13,23. La radioterapia no pudo ser catalogada como factor de riesgo en pacientes con MPNST ni mostró mejoría estadísticamente significativa en la ST4,23. Sorprendentemente, en otro estudio, los pacientes que recibieron radioterapia, con dosis menores a 45 Gy, tuvieron una mayor frecuencia de recurrencia que quienes no la recibieron18. Al igual que con la quimioterapia, se requerirá de estudios prospectivos aleatorizados para concluir sobre el beneficio de este tratamiento.
En vista de los regulares resultados de SLE y ST con los tratamientos convencionales, y teniendo en cuenta las alteraciones cromosómicas encontradas en muchos de los MPNST, los investigadores han dirigido sus trabajos hacia el uso de sustancias diana que pueden actuar eliminando células tumorales con menos efectos secundarios. Se sabe que el gen responsable de la NF1 está ubicado en el cromosoma 1724 y codifica para la neurofibromina, localizada en 17q11.225, que funciona como un gen supresor de tumor expresado fuertemente en tejido neural, riñón, bazo y hueso24 e interactúa con el gen Ras, manteniéndolo en forma inactiva25. Las mutaciones de NF1 que generan una pérdida de la expresión de la neurofibromina se consideran eventos promotores de tumor, ya que se activa Ras, oncogén responsable de proliferación, invasión y metástasis de las células tumorales25. La mayoría de las vías de transducción de señales intracelulares que llevan a activación de Ras y otras proteínas promotoras de tumor se originan por receptores de tirosina cinasa ubicados en la membrana celular. Por lo tanto, la inhibición de estos receptores con medicamentos antagonistas disponibles en el mercado (como imatinib, dasatinib, sunitinib, sorafenib, etcétera) puede ser de importancia en el tratamiento25, aunque los resultados preliminares aún no son muy alentadores. Sin embargo, ya es posible identificar candidatos para la terapéutica molecular dirigida. Alaggio y colaboradores26 estudiaron el gen BIRC5/SURVIVINA, en la región cromosómica 17q de células tumorales en pacientes con MPNST, y encontraron que sus valores elevados de correlacionan significativamente con la extensión del tumor y con la probabilidad de sobrevida, lo que apoya el concepto de que la SURVIVINA puede ser considerada como un marcador pronóstico y un objetivo para intervenciones terapéuticas. De igual manera, Patel y colaboradores27 trabajando en células de tumores MPNST de ratones y de humanos, identificaron la sobreexpresión y amplificación del gen AURKA (Aurora Kinasa), cuyo objetivo es Ras, y demostraron que al bloquear este gen disminuía el crecimiento de las células tumorales in vivo e in vitro.
Finalmente, las histonas deacetilasas (HDAC) son una familia de enzimas involucradas en la expresión de genes, reparación de DNA y respuesta al estrés; todos estos procesos se alteran en tumores y por lo tanto los inhibidores HDAC tienen alta actividad antitumoral28. La inhibición de la isoforma HDAC8 induce detención del crecimiento en la fase S del ciclo celular29, por lo que el uso de sustancias farmacológicas que inhiban esta enzima —en combinación con agentes quimioterapéuticos como algunos antimetabolitos (como 5-fluorouracilo, gemcitabina y citarabina) que fundamentalmente actúan sobre la fase S—, son procedimientos muy prometedores para obtener un mejor resultado en el control de tumores como los MPNST28,29. Diferentes estudios confirman que las HDCA son excelentes objetivos en el tratamiento del cáncer; son varios los inhibidores HDAC que han demostrado efectividad en un amplio rango de enfermedades hematológicas y tumores sólidos, disminuyendo notoriamente la toxicidad30.
Los MPNST son altamente malignos, con una tasa de recidiva entre 40-65% y una tasa de metástasis entre 40-68%, las cuales dependen del grado de malignidad histológica y le confieren un pronóstico pobre3. Los sitios de metástasis principalmente son pulmón, hueso y pleura en orden de frecuencia, por lo que la radiografía de tórax es útil como se indicó en nuestra paciente; sin embargo, se prefieren la TAC de tórax y la gammagrafía ósea dentro de la evaluación de enfermedad metastásica6.
Aproximadamente 25-50% de los MPNST ocurren en pacientes con NF1, y otro 10- 20% ocurren después de irradiación ocupacional o terapéutica31. La paciente reportada se trata de un caso esporádico, pues no cumple los criterios clínicos ni tiene historia familiar de NF1 ni ha recibido radioterapia. En el trabajo de Anghileri y colaboradores32, se logró establecer que los principales factores pronóstico que influyen en la sobrevida son la recurrencia de la enfermedad, el tamaño del tumor y su localización (tronco o extremidades). Adicionalmente, los pacientes que tuvieron márgenes quirúrgicas positivas tuvieron 2.4 veces más riesgo de desarrollar recaída local, y el 50% de los pacientes que recibieron radioterapia presentaron más frecuentemente recaídas locales, aunque no hubo diferencia estadística con quienes no la recibieron32. De manera interesante, la presencia de NF-1 no afectó la sobrevida como factor independiente, dato corroborado en el meta-análisis publicado por Kolberg y colaboradores33, pero los pacientes con NF-1 tuvieron tumores de mayor tamaño y aparición más temprana de los mismos32. La paciente que presentamos en este reporte tiene dos factores de riesgo muy importantes: el tamaño tumoral y la presencia de tumor residual postquirúrgico. Independientemente de los factores pronósticos clínicos, investigadores noruegos demostraron alteraciones en los cromosomas 10, 16 y X de células tumorales de pacientes con MPNST. Ellos encontraron que ganancias en 16p, o pérdidas de 10q o Xq, identificaban un grupo de muy alto riesgo que solo alcanzaba una sobrevida de 11% a 10 años con diferencia estadística altamente significativa (p < 0.00005)34.
A pesar de la terapia multimodal, la ST a los 5 años oscila entre 35-50%22,35. En la serie pediátrica ítalo-alemana se encontró una ST de 43-59%4.
A pesar de su baja incidencia, los MPNST son importantes debido a su agresividad, y deben sospecharse ante una masa localizada a nivel paravertebral o en extremidades, en especial en pacientes con NF1. La piedra angular en el tratamiento es la resección quirúrgica completa debido a la alta tasa de recidiva, con una respuesta terapéutica limitada a la radioterapia y quimioterapia. Este caso muestra las manifestaciones clínicas y las complicaciones que se pueden esperar con estos tumores, así como su comportamiento agresivo. La ausencia de NF1 no descarta el diagnóstico.
Responsabilidades éticas
Protección de personas y animales
Los autores declaran que para esta investigación no se han realizado experimentos en seres humanos ni en animales.
Confidencialidad de los datos
Los autores declaran que han seguido los protocolos de su centro de trabajo sobre la publicación de datos de pacientes.
Derecho a la privacidad y consentimiento informado
Los autores han obtenido el consentimiento informado de los pacientes y/o sujetos referidos en el artículo. Este documento obra en poder del autor de correspondencia.
Financiamiento
Recursos propios de los autores.
Conflicto de intereses
Los autores declaran no tener ningún conflicto de intereses.

