










Servicios Personalizados
Revista
Articulo
 Español (pdf)
Español (pdf)
 Artículo en XML
Artículo en XML Referencias del artículo
Referencias del artículo
 Enviar artículo por email
Enviar artículo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO -
 Accesos
Accesos
Links relacionados
-
 Similares en
SciELO
Similares en
SciELO
Compartir

 Permalink
PermalinkBoletín médico del Hospital Infantil de México
versión impresa ISSN 1665-1146
Bol. Med. Hosp. Infant. Mex. vol.71 no.5 México sep./dic. 2014
Artículo de revisión
Neoplasias malignas en el neonato
Malignant neoplasms in the neonate
Marta Zapata-Tarrésa,*, Daniel Ibarra-Ríosb, Irma Viridiana Cruz-Rodríguezc, Luis Enrique Juárez-Villegasd, Humberto Peña-del Castilloe
a Servicio de Oncología, Instituto Nacional de Pediatría, México D.F., México.
b Departamento de Neonatología, Hospital Infantil de México Federico Gómez, México D.F., México.
c Medicina Materno Fetal, Hospital Gineco-Obstetricia Luis Castelazo Ayala, México D.F., México.
d Servicio de Oncología, Hospital Infantil de México Federico Gómez, México D.F., México.
e Enseñanza, Instituto Nacional de Pediatría, México D.F., México.
Autora para correspondencia:
M. Zapata-Tarrés
Correo electrónico: magazapata@yahoo.com
Recibido el 23 de mayo de 2013;
aceptado el 30 de mayo de 2014.
Resumen
El cáncer en la edad pediátrica presenta características que lo diferencian de otros tipos reportados en edades posteriores. La supervivencia global a 3 años es de hasta el 70%, dependiendo de la neoplasia estudiada. Los principales aparatos y sistemas afectados son el sistema hematopoyético, el sistema nervioso central y simpático, así como tejidos mesenquimatosos.
El incremento en la incidencia de tumores neonatales observado en este y otros estudios se basa en el aumento del número de tumores sólidos (teratomas y neuroblastomas), ya que los casos de tumores en el sistema nervioso central y leucemias han permanecido constantes.
La ultrasonografía es la primera línea de abordaje y puede detectar hasta el 70% de las anomalías fetales.
La fisiología del neonato hace que el tratamiento multidisciplinario necesario en las enfermedades neoplásicas sea modificado sustancialmente en este grupo de edad, para evitar toxicidad y secuelas. El tratamiento más utilizado es la cirugía.
Logrando el diagnóstico oportuno existen opciones terapéuticas efectivas para mejorar la supervivencia de estos pacientes.
Palabras clave: Cáncer; Tumores neonatales; Neoplasias; Tratamiento.
Abstract
Cancer in children has characteristics that differentiate it from other types reported in later ages. Overall survival at 3 years is up to 70% depending on the tumor studied. Major organs and systems affected are the hematopoietic system, central nervous system and sympathetic and mesenchymal tissues.
The increased incidence of neonatal tumors observed in this and other studies is based on the increasing number of solid tumors (teratomas and neuroblastomas) because cases of central nervous system tumors and leukemias have remained constant.
Ultrasonography is the first line of approach and can detect up to 70% of fetal anomalies.
The physiology of the newborn causes the necessary multidisciplinary treatment in neoplastic disease to be modified substantially in this age group to avoid toxicity and sequelae. The most common treatment is surgery.
Achieving timely diagnostic treatment options are effective in improving the survival of these patients.
Keywords: Cancer; Neonatal tumors; Neoplasms; Treatment.
1. Introducción
El cáncer que se desarrolla en la edad pediátrica posee características propias que lo diferencian de aquellos presentes en edades posteriores de la vida, incluso en el pronóstico: se reporta una supervivencia global a 3 años de hasta el 70% que varía de acuerdo con el tipo de neoplasia estudiada. Los principales aparatos y sistemas afectados son el sistema hematopoyético, el sistema nervioso central y simpático, así como tejidos mesenquimatosos1,2.
Dentro del grupo de neoplasias pediátricas existe un subgrupo que merece mención especial. Estos son los tumores neonatales que se presentan durante las cuatro primeras semanas de vida extrauterina. En la literatura mundial se emplean distintos términos que, si bien definen subgrupos distintos, son utilizados con frecuencia como sinónimos (tabla 1)3.

Aunque los tumores neonatales son un grupo de neoplasias raras, representan un gran blanco de atención de los investigadores, ya que aún son un enigma respecto de su etiología, diagnóstico, pronóstico y tratamiento. Cabe mencionar que difieren en incidencia, grado de diferenciación histológica, respuesta terapéutica (aumento de la toxicidad por quimioterapia y mayor número de complicaciones quirúrgicas) y pronóstico que, además de la inmadurez anatómica y fisiológica propia de la edad del paciente y característica del periodo neonatal, requieren de terapéuticas y métodos diagnósticos especiales4.
2. Epidemiología
Las neoplasias en el grupo pediátrico constituyen el 2% del total de las mismas. De estas, el 17% corresponde a tumores diagnosticados durante la etapa neonatal, con una incidencia de 2.8 a 3.74 casos por 100,000 recién nacidos vivos por año. De los 7,000 niños diagnosticados por año con cáncer en EE. UU., el 10% se diagnostica durante el primer año de vida, el 2% durante el primer mes y solamente el 1% en el primer día de vida extrauterina. Según lo reportado por Vasilatau-Kosmidis en 2003, las neoplasias congénitas correspondieron a uno de cada 12,500 nacidos vivos, similar a lo publicado en otros estudios, con diferente distribución de los tipos de cáncer de acuerdo con la edad de los pacientes (tabla 2) e incidencia de distintos tumores neonatales (tabla 3)5,6.


Según datos del National Cancer Institute's Surveillance Epidemiology and End Results Program (SEER), la incidencia de cáncer en menores de 12 meses muestra un incremento año tras año desde 1970 hasta del 15%. Han existido variaciones en la distribución por sexos, ya que los pacientes del sexo masculino han presentado un aumento de hasta el 30% a expensas de tumores de sistema nervioso central (SNC), retinoblastomas y neuroblastomas, mientras que en mujeres solamente han aumentado el 6%, tomando como base los tumores hepáticos7. En el Reino Unido, entre 1993 y 2007, se reportaron 303 neonatos con cáncer durante el primer mes de vida. Esto da como resultado una incidencia de 28 neonatos con cáncer por millón de recién nacidos vivos (fig. 1)8. En la tabla 4 se muestra la experiencia de 69 años en el Hospital Infantil de México Federico Gómez9.


El incremento en la incidencia de tumores neonatales observado en este y otros estudios se basa en el aumento de tumores sólidos (teratomas y neuroblastomas). El número de tumores en el SNC así como los casos de leucemias han permanecido constantes10-16. El aumento en el diagnóstico de estos procesos patológicos está relacionado con el incremento del uso rutinario de técnicas de imagen durante el embarazo, como la ecografía fetal, así como de la aplicación de métodos diagnósticos de tamización17-19. Un claro ejemplo de esto se observa en Japón y Canadá, donde se ha estandarizado la toma de catecolaminas para el diagnóstico de neuroblastoma20-23.
3. Diagnóstico prenatal
Con el desarrollo de la perinatología y la medicina maternofetal, hoy en día es posible la detección prenatal de muchos tumores, tanto abdominales como mediastínicos y de SNC24. La ultrasonografía es la primera línea de abordaje y puede detectar hasta el 70% de las anomalías fetales25. El siguiente paso en el diagnóstico prenatal es la resonancia magnética fetal. La ecocardiografía fetal con Doppler también es muy útil para determinar malformaciones cardiacas y estados hemodinámicos secundarios a la actividad de un tumor o por secuestro de volumen. Puede tener un papel importante en tumores intracardiacos. Con una cordocentesis se puede obtener sangre fetal para análisis cromosómico y para otras mediciones. También, con una amniocentesis y biopsia de vellosidades coriónicas se puede realizar cariotipo, buscar microarreglos cromosómicos y mutaciones específicas.
En los tumores de SNC es frecuente encontrar polihidramnios secundario a disfunción hipotalámica, lo que disminuye la deglución de líquido amniótico. La masa en cuestión se localiza por ultrasonido o se encuentran datos indirectos, como hidrocefalia secundaria a la compresión. En uno de cada cinco tumores puede haber hemorragia.
Los tumores en cara y cuello tienen un amplio diagnóstico diferencial. Se incluyen teratomas, linfangiomas, bocio congénito, tumores tiroideos, tumores tímicos, neuroblastomas y hamartomas. Las lesiones más comunes que adquieren un tamaño sustancial son los teratomas. Es importante su detección y medición porque permiten planear la resolución del embarazo en un centro de atención de tercer nivel. El compromiso de la vía aérea puede poner en riesgo la vida. Se ha descrito el procedimiento EXIT en el cual se realiza una cesárea y se accede a la vía aérea mientras el feto está bajo circulación placentaria. Si se realiza adecuadamente, la circulación placentaria permite hasta 2 h para realizar laringoscopia, broncoscopia, traqueostomía, e incluso la resección tumoral26.
Los tumores cardiacos se pueden encontrar durante la revisión de rutina, y además acompañarse de cardiomegalia, derrame pericárdico, arritmias o falla cardiaca.
Cuando se encuentran lesiones solitarias, se debe pensar en rabdomiomas, teratomas o fibromas. Se espera que en un paciente con rabdomiomas múltiples, estos involucionen con el tiempo. Sin embargo, debe hacerse un seguimiento estrecho por el riesgo de tumores de SNC al encontrar una probable esclerosis tuberosa27.
Cuando se observan tumores mediastínicos generalmente se piensa en teratomas, pero también pueden ser quistes tímicos, bocio intratorácico, neuroblastoma o duplicaciones intestinales.
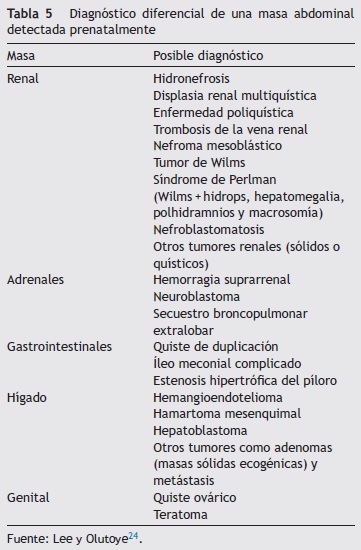
En caso de masas pulmonares se debe determinar de manera posnatal la histología, para diferenciar malformaciones adenomatoideas quísticas, secuestro broncopulmonar, atresia bronquial, enfisema lobar congénito o, más raramente, blastoma pleuropulmonar o tumor de tejido intersticial. En la tabla 5 se muestra el diagnóstico diferencial de una masa abdominal detectada prenatalmente.
En el caso de las leucemias existen datos ultrasonográficos, hepatoesplenomegalia e hidrops fetal así como polihidramnios, que ayudan a la sospecha de leucemia antenatal, corroborando el diagnóstico por medio de fetoscopia y cordocentesis (extrayendo una muestra de sangre del cordón y haciendo una tinción Wright-Giemsa). La anemia fetal puede diagnosticarse y vigilarse mediante Doppler de la arteria cerebral media. Hay reportes de fetos con síndrome de Down con diagnóstico prenatal de leucemia a la semana 33 de gestación.
En padecimientos oncológicos con orígenes genéticos, como el caso del retinoblastoma, existe hoy en día la posibilidad de realizar un diagnóstico de preimplantación, analizando los embriones y realizando PCR para detectar la mutación del gen RB1. De esta manera se localizan embriones no afectados y se transfieren al útero al día 528,29.
Para la detección de neuroblastoma en países como Japón y Canadá, se trató de cuantificar el número de pacientes a través de la tamización neonatal con la medición los metabolitos de las catecolaminas en orina30. Se encontró que aproximadamente la mitad de todos los neuroblastomas que alcanzarían un tamaño detectable por tamización tendrían una regresión espontánea. También se concluyó que no es recomendable la tamización por el costo-beneficio; sin embargo, en Japón está aún en discusión.
4. Dificultades en el diagnóstico de los tumores neonatales
Generalmente, los patólogos establecen el diagnóstico de enfermedad maligna basándose en criterios histológicos, los cuales no necesariamente son extrapolables a los pacientes pediátricos. Esto sucede especialmente en los tumores que se presentan en los primeros 28 días de vida extrauterina. Normalmente, el desarrollo de los órganos y tejidos muestra un aumento de la actividad mitótica, y tiene apariencia de estructuras inmaduras que podrían confundirse con neoplasias malignas. Además, durante el periodo neonatal, las características histológicas de un tumor no son siempre indicativas del comportamiento biológico del mismo ni de su evolución. Particularmente en este grupo etario, la localización desempeña un papel preponderante en la morbimortalidad del paciente ya que hay tumores que, a pesar de no ser malignos histológicamente, ocasionan la muerte debido al lugar donde se originan, así como las estructuras adyacentes que afectan. Cabe señalar que existe un sinnúmero de procesos patológicos que pueden simular un cuadro tumoral, y que son las mismas de características benignas en estos pacientes. Ejemplos de esto son las enfermedades involucradas en el sistema gastrointestinal (atresia intestinal, malformación anorrectal, obstrucciones meconiales, fibrosis quística, lifangioma, etcétera), hasta en el 15%; las genitourinarias (hidronefrosis, hidrouréter, quiste de uraco, divertículo de uraco, riñón multiquístico, poliquistosis renal, trombosis venosa renal, hidrocolpos, quiste ovárico, torsión ovárica, etcétera), en el 55%; las adrenales (5%) y las hepatobiliares (5%), entre otras.
De la misma forma, algunas neoplasias malignas pueden presentar un comportamiento benigno, como el caso del neuroblastoma, incluso cuando se observa involución espontánea31.
5. Etiopatogenia de los tumores neonatales
La etiopatogenia de los tumores neonatales difiere a la reportada en otras etapas de la vida1,3. En la población general, las neoplasias aparecen durante la segunda mitad de la vida, ya que se requiere de largos periodos de exposición y latencia a agentes cancerígenos (físicos, químicos o biológicos) para producir mutaciones en los genes supresores tumorales, así como en los protooncogenes32-34. A diferencia de los adultos, en la edad pediátrica existe una mayor susceptibilidad a estos agentes, condicionada por una mayor división celular con un menor tiempo de reparación del ADN y una mayor proliferación clonal; una menor actividad reparadora mutacional; una inmadurez fisiológica en los mecanismos hormonales, de detoxificación e inmunovigilancia; y una mayor predisposición a los agentes cancerígenos en la inducción de anomalías en el desarrollo35-37.
Como se mencionó anteriormente, los tumores neonatales se componen de tejido embrionario que se mantiene sin cambio a través del tiempo, lo que hace pensar que su génesis puede deberse a fallas en el mecanismo de maduración durante las primeras etapas de la vida. Dentro de los principales mecanismos de oncogénesis descritos en los tumores neonatales se encuentran los preconcepcionales, donde los factores ambientales (radiaciones ionizantes, no ionizantes, agentes infecciosos, fármacos y drogas, tabaco, alcohol, dieta y ocupaciones parentales) pueden actuar sobre las células germinales de los progenitores alterando las mismas38. Estos agentes afectan todas las células del organismo, incluidas las germinales (óvulos y espermatozoides), produciendo alteraciones genéticas previas a la concepción, las cuales favorecen la aparición de neoplasias en sus descendientes en un periodo variable de tiempo, desde el periodo neonatal hasta la edad adulta. En el hombre, la espermatogénesis comienza en la pubertad y finaliza a edad avanzada, existiendo un largo periodo de exposición a los diferentes agentes cancerígenos. Por otro lado, en las mujeres, los óvulos se forman durante su gestación, por lo que se detiene la creación de nuevas células germinales una vez que han nacido, lo que condiciona un corto periodo en el que puedan actuar los agentes cancerígenos. Por ello, cuando los estudios epidemiológicos intentan relacionar las diferentes exposiciones ambientales, y más concretamente las relativas a ocupaciones parentales, con los tumores pediátricos, es en los padres y no en las madres donde se obtienen resultados positivos39.
Un tipo especial de oncogénesis preconcepcional es la transgeneracional, que da origen a los cánceres hereditarios o familiares en los que la o las mutaciones se heredan con carácter dominante o recesivo, incrementando de manera significativa la aparición de neoplasias de manera precoz, con mayor frecuencia de lesiones en el mismo órgano, afección bilateral en órganos pares y la presencia simultánea de múltiples cánceres primarios40.
De acuerdo con el momento en que afecta las células, la oncogénesis preconcepcional se puede dividir, primeramente, en transplacentaria, donde las alteraciones ocurren durante el tiempo que dura la gestación e implica necesariamente el paso de sustancias mutagénicas a través de la barrera placentaria hacia el feto, generándose una neoplasia posterior en un periodo variable de tiempo (incluso hasta la edad adulta)41,42. Determinados agentes tóxicos, mutagénicos y cancerígenos transplacentarios originan efectos diferentes según la fase de la gestación en la cual actúen: abortos (en las primeras semanas), malformaciones (entre las semanas 2 y 8) o tumores (entre las semanas 6 y 40).
La segunda etapa corresponde a la de transmisión materno-fetal de células tumorales. En este caso no se da una transmisión madre-hijo de las mutaciones como tal. En la gran mayoría, la placenta actúa como una barrera que previene el paso de metástasis de células tumorales de la madre al feto y viceversa. Excepcionalmente se han descrito metástasis de este tipo en melanomas, coriocarcinomas, linfomas, carcinomas broncogénicos y epiteliomas mamarios.
Por último, la etapa de presentación posnatal, donde el corto tiempo de exposición y latencia hace pensar que durante esta etapa no existe oncogénesis.
Otra característica muy peculiar de los tumores neonatales es el término utilizado por primera vez por Bolande en 1985, conocido como «periodo de gracia oncológico neonatal»35. El mismo se refiere a que la mayoría de los tipos histológicos en los tumores neonatales presentan un comportamiento biológico menos agresivo que en épocas posteriores de la vida, por lo que la edad per se es un factor pronóstico importante e independiente del tipo tumoral, patrón histológico y extensión del mismo. Esto se debe a dos fenómenos diferentes que condicionan una desaparición parcial o completa del tumor: la regresión espontánea (desaparición total de las células tumorales) y la citodiferenciación (maduración celular maligna o benigna).
5.1 Factores de riesgo
Las neoplasias se originan por la combinación variable de dos tipos de determinantes, el genético y el ambiental. Los factores ambientales son los responsables del 98% de todos los cánceres. Los agentes cancerígenos se clasifican en químicos, físicos y biológicos. Para el efecto de los mismos, se requiere de largos periodos de latencia (desde un par de años hasta décadas) para desarrollar las neoplasias. En esta variabilidad influyen al menos dos factores: primero, la mayor o menor susceptibilidad genética para desarrollar el cáncer; y segundo, que se necesitan como mínimo 5-6 mutaciones de los protooncogenes y genes supresores tumorales para la transformación maligna de una célula. La mayoría de los cánceres pediátricos se desarrollan tras breves periodos de tiempo. Hasta el 40% se presentan durante los primeros 4 años de vida. La mayoría de los autores asocian los factores genéticos con el 4-10% de los tumores infantiles. Los casos restantes son responsabilidad de los agentes ambientales, explicando el corto periodo de latencia por sus acciones durante el embarazo y previo a este sobre tejidos celulares morfológica y funcionalmente inmaduros43,44.
5.2 Factores genéticos asociados con cáncer pediátrico
Los síndromes genéticos son la causa del 1-2% de todos los cánceres, con una mayor frecuencia en la edad pediátrica (4-15%). Estrictamente, se denominan cánceres hereditarios o genéticos los que se desarrollan en pacientes portadores de mutaciones específicas de las células germinales y que, por lo tanto, están presentes en todas las restantes células somáticas. Dichos procesos patológicos se pueden dividir, de acuerdo con la forma de herencia, en recesivos (ataxia telangiectasia, anemia de Fanconi, xeroderma pigmentosum, síndrome de Bloom), dominantes (síndrome de Li-Fraumeni, retinoblastoma familiar, neurofibromatosis, tumor de Wilms familiar, neuroblastoma familiar, síndrome de neoplasia múltiple endocrina, esclerosis tuberosa, síndrome de von Hippel-Lindau) o cromosómicos no hereditarios (síndrome de Down y alteraciones de los cromosomas sexuales)1.
5.3 Factores ambientales asociados con cáncer pediátrico
Varios factores ambientales se han intentado asociar con el cáncer en la edad pediátrica, aunque los resultados de los estudios realizados son controvertidos. Entre los más estudiados se encuentran la radiación ionizante1,45-48, la radiación no ionizante49-53, infecciones54, fármacos, dieta55,56, tabaquismo57, alcoholismo58,59 y exposición a ocupación de los padres (sobre todo la exposición paterna a los pesticidas, solventes, pinturas y empleos relacionados con los vehículos de motor se han asociado con un mayor número de tumores a nivel de SNC)60,61.
6. Tratamiento de los tumores neonatales
La fisiología propia del neonato hace que el tratamiento multidisciplinario necesario en las enfermedades neoplásicas sea modificado sustancialmente, para evitar la toxicidad y secuelas62,63. El tratamiento más utilizado es la cirugía, lo que requiere de una preparación y estabilización preoperatoria así como avanzados cuidados postoperatorios31. Le sigue, en frecuencia, la quimioterapia, la cual requiere modificar la dosis y las vías de administración de los diversos fármacos quimioterapéuticos. La absorción, biotransformación y excreción de estos fármacos difieren de las de otras edades. Como ejemplo tenemos que la disminución del pH gástrico prolonga el vaciado gástrico, y los constantes cambios en las resistencias vasculares mesentéricas y periféricas hacen variable el flujo sanguíneo, modificando la absorción y distribución de los fármacos, ya sea por vía oral o intravenosa. La inmadurez hepática y renal altera la tasa de eliminación de los diversos agentes quimioterapéuticos (metotrexato, cisplatino y ciclofosfamida; y vincristina y actinomicina D, respectivamente); y las proteínas plasmáticas, especialmente la albúmina, que normalmente transportan y modulan la toxicidad de la quimioterapia (metotrexato, 6-mercaptopurina, prednisona), suelen estar saturadas por la mayor afinidad y exceso de bilirrubina y ácidos grasos libres, alterándose la distribución y aumentando la toxicidad64. El relativo aumento del volumen extracelular y los excesos de grasa en el recién nacido producen un incremento de la biodisponibilidad de muchos agentes quimioterapéuticos. Para mantener la efectividad y minimizar la toxicidad, algunos autores y centros médicos especializados utilizan la mitad de la dosis habitualmente recomendada en pediatría y la calculan por kilogramo de peso (en vez de su cálculo clásico por superficie corporal). La radioterapia normalmente se omite ya que lesiona gravemente los tejidos sanos, ocasionando serios problemas a corto plazo así como secuelas por deformidad, además del mayor poder oncogénico de la misma36.
7. Diferentes tipos de cáncer neonatal65
7.1 Tumores germinales fetales y neonatales
Los tumores de células germinales son un reto debido a que su pluripotencialidad intrínseca resulta en un amplio espectro de histologías y comportamiento biológicos. Representan actualmente (en competencia con el neuroblastoma) el tipo más común de tumor neonatal. Debido a su tamaño, localización y complejidad, el manejo óptimo requiere un equipo multidisciplinario de obstetras, neonatólogos, cirujanos, radiólogos y oncólogos66. El 95% de los tumores de células germinales están compuestos de teratomas maduros o inmaduros, curables únicamente con cirugía. La alfafetoproteína es un marcador confiable para detectar la recurrencia de la enfermedad para iniciar el manejo adecuado (tabla 6). Hay controversia sobre la necesidad de bordes quirúrgicos negativos ya que, muchas veces, pueden ser difíciles de lograr, y las recaídas no son tan frecuentes (incluso en etapa 2). Si es necesaria la quimioterapia, se debe dar un esquema basado en carboplatino, ajustado y minimizado para evitar toxicidad. En el raro caso del neonato con enfermedad metastásica, la conducta a seguir es la quimioterapia neoadyuvante tras la biopsia que da el diagnóstico.

7.2 Neuroblastoma
El segundo, y en algunas series el cáncer más frecuente en el periodo neonatal, es el neuroblastoma67. Tiene, en general, un pronóstico bueno, sobre todo en aquellos casos en los que no hay amplificación del oncogén MYC. Los casos en los que sí hay amplificación (cualquiera que sea el estadio) deben ser tratados como pacientes de alto riesgo con un régimen intensivo de quimioterapia. Clínica y radiológicamente, la presentación es similar a la de otras edades, y se observan síntomas relacionados con la producción de catecolaminas y presencia de calcificaciones a nivel radiológico. El diagnóstico oportuno y el tratamiento urgente son fundamentales en los casos que se presentan con compresión medular por una masa paraespinal, o coagulopatía por infiltración hepática en la enfermedad metastásica. La enfermedad metastásica puede tener regresión espontánea en este grupo etario, por lo que muchas veces se maneja con observación. El abordaje quirúrgico debe ser conservador y realizarse al existir factores de riesgo bien establecidos. Ante la presencia del síndrome de Pepper (tumor primario localizado con metástasis limitadas a hígado, piel y médula ósea) existe alta mortalidad en esta etapa de la vida.
7.3 Tumores de sistema nervioso central
Actualmente ha aumentado el diagnóstico prenatal de estos tumores. Los síntomas neonatales difieren de otras etapas de la vida ya que, al estar las suturas craneales abiertas, se observa que el síntoma o signo más frecuente es el aumento del perímetro cefálico sin hipertensión intracraneal secundaria. La hidrocefalia debe considerarse un signo de alarma. En los neonatos son raros los datos neurológicos focales. Los teratomas son el tipo histológico más frecuente, seguido por los astrocitomas, meduloblastomas, tumores de plexos coroideos y craneofaringiomas. Los tumores de plexos coroideos (generalmente papilomas) tienen el mejor pronóstico, y en algunos casos la cirugía agresiva es curativa. El resto de las histologías tienen un pronóstico pobre, aunque algunas estirpes podrían responder a cirugía agresiva y quimioterapia. Es necesaria la participación de un equipo multidisciplinario ya que, por ejemplo, son frecuentes las complicaciones neuroendocrinas68,69.
7.4 Leucemia neonatal
La leucemia neonatal (o congénita) es aquella diagnosticada en los primeros 30 días a partir del nacimiento70. Su incidencia es de 1-5 por cada millón de recién nacidos vivos (menos del 1% del total de las leucemias). La experiencia de varios países en periodos de 20 a 50 años comprende únicamente decenas de casos. La literatura internacional reporta leucemias mieloides del 56-64% y linfoides del 21-38%. El 50% de las leucemias mieloides son monoblásticas (M5). Dentro de la clínica para el diagnóstico, se encuentra hepatoesplenomegalia en el 80%, hiperleucocitosis en el 85% y leucemia cutis en el 60%. En todos los pacientes debe hacerse el diagnóstico diferencial para infecciones por el complejo TORCH, incompatibilidad a grupo, otros cánceres, como neuroblastoma e histiocitosis de células de Langerhans, etcétera. En la literatura internacional se ha descrito una supervivencia de menos del 10% para las linfoides y del 25% para las mieloides.
7.5 Síndrome mieloproliferativo transitorio del recién nacido asociado con síndrome de Down
Es una proliferación descontrolada de mieloblastos que, en ocasiones, puede ser difícil diferenciar de la leucemia neonatal o congénita, aunque este síndrome tiende a resolverse espontáneamente dentro de los primeros 3 meses de vida. Es generado por mutaciones en el gen codificador de la proteína GATA 1 al producir una proteína truncada, ya que altera la función reguladora de la megacariopoyesis71. La mayoría de los neonatos no requieren tratamiento.
7.6 Tumores renales
El tipo de tumor más común es el nefroma mesoblástico congénito (60%), que puede ser confundido con tumor de Wilms. El nefroma mesoblástico no está encapsulado e infiltra el parénquima renal normal. Debe ser resecado quirúrgicamente, ya que se considera que puede progresar a tumor de Wilms si no es extirpado completamente. Incluso, se han reportado casos de metástasis de este tumor. En segundo lugar se encuentra el tumor de Wilms (20%), seguido por el tumor rabdoide (11%) y los sarcomas de células claras. Estos tumores tienen buen pronóstico en manos de un oncólogo, un cirujano oncólogo y un radioterapeuta expertos por lo que, cuando se sospeche de un tumor así, debe ser referido inmediatamente14,72.
7.7 Tumores hepáticos
Los más frecuentes son los hemangioendoteliomas. Estos tumores muchas veces se acompañan de lesiones cutáneas similares, que típicamente involucionan tras el año. A su vez, esta asociación se relaciona con hipotiroidismo, como resultado de la triyodotironina deiodinasa en estos tumores. Las lesiones asintomáticas se manejan de manera conservadora. En lesiones con repercusión hemodinámica se administra tratamiento con propranolol, corticoesteroides y vincristina. El hepatoblastoma representa solamente el 1% de las neoplasias pediátricas12,13. De este porcentaje, aproximadamente el 10% ocurre en el periodo neonatal, y son más frecuentes en prematuros de bajo peso. El tratamiento incluye resección quirúrgica, con o sin quimioterapia previa, y consolidación con quimioterapia. La alfafetoproteína es un marcador de esta neoplasia (tabla 6)73.
7.8 Retinoblastoma
Es el tumor maligno intraocular más frecuente. Se desconoce la incidencia real en el recién nacido. Puede ser esporádico o hereditario. Los casos unilaterales pueden ser esporádicos o hereditarios, mientras que los casos bilaterales son siempre hereditarios. El 90% de los casos son esporádicos. El síntoma más común es la leucocoria, seguido de estrabismo, inflamación de la órbita, hifema, pupila fija y heterocromía del iris. Es importante mencionar que, en el periodo neonatal, el estrabismo y la inflamación de la órbita pueden aparecer como alteraciones «normales», por lo que es obligatorio un examen del fondo de ojo con dilatación pupilar al nacimiento. La biopsia diagnóstica está estrictamente prohibida por el riesgo de esparcir el tumor. Se deben evitar las tomografías computadas por el riesgo de segundas neoplasias. El tratamiento incluye muchas modalidades, como quimioterapia sistémica, láser, crioterapia y braquiterapia. La enucleación se ofrece cuando hay pocas posibilidades de salvar el ojo74.
7.9 Sarcomas de tejidos blandos
Los tumores vasculares son los más comunes, y generalmente son benignos, aunque pueden ocurrir complicaciones que ponen en riesgo la vida. Los tumores benignos pueden tener apariencia clínica y radiológica similar a los malignos, por lo que el diagnóstico diferencial puede ser muy difícil. El fibrosarcoma infantil es típico de la etapa neonatal, y se considera un tumor de pronóstico intermedio. Frecuentemente se ofrece quimioterapia neoadyuvante libre de agentes alquilantes y antraciclinas para reducir el tumor para su posterior resección quirúrgica. El rabdomiosarcoma es un cáncer grave que requiere manejo multidisciplinario. El pronóstico en la etapa neonatal es pobre75.
8. Pronóstico
Por todo lo anterior, el hecho de presentar un tumor en el periodo neonatal no es sinónimo de muerte ya que, además de los casos que responden de manera adecuada al tratamiento quirúrgico o con quimioterapia, hay tumores que presentan una regresión espontánea o maduración (citodiferenciación). La tasa de supervivencia ha mejorado con el tiempo. Los pacientes con neuroblastoma registran la supervivencia más alta; sin embargo, casi todos los pacientes con diagnóstico de tumores cerebrales y leucemias fallecen5.
Las neoplasias en la etapa neonatal representan un grupo aparte del resto de las presentadas en la edad pediátrica y adulta por sus peculiaridades en incidencia, grado de diferenciación histológica, respuesta terapéutica y pronóstico. Para su diagnóstico, es necesaria la experiencia tanto de los médicos oncólogos pediatras como de todo el equipo diagnóstico (incluidas otras especialidades) y, por supuesto, de personal de patología con extenso conocimiento en tumores e histología neonatal. El diagnóstico representa un desafío debido a la compleja anatomía y fisiología del neonato. Desde un punto de vista patobiológico, los tumores neonatales representan importantes modelos para el estudio de la oncogénesis humana, y ofrecen información muy importante respecto de la fisiopatología del cáncer en cuanto a genética y patrón hereditario.
Aunque la oncogénesis no esté dilucidada por completo, existen importantes asociaciones con grados variables de correlación, así como factores de riesgo que se deben admitir para crear la posibilidad diagnóstica de estos procesos patológicos, forjando la conciencia médica de la existencia de tumores malignos en la etapa neonatal. Cuando se logra el diagnóstico oportuno, existen opciones terapéuticas efectivas para mejorar la supervivencia de estos pacientes. Al ser padecimientos raros, se requiere de la experiencia acumulativa de muchos centros de atención terciaria para acercarse al mejor manejo en estos pacientes.
Conflicto de intereses
Los autores declaran no tener ningún conflicto de intereses.
Referencias
1. Ferrís i Tortajada J, García i Castell J, López-Andreu-JA, Pellicer-Porres C. Factores genéticos asociados a cánceres pediátricos. An Esp Pediatr. 1999;50:4-13. [ Links ]
2. Devessa SS, Blot WJ, Stone BJ, Miller BA, Tarone RE, Fraumeni FR Jr. Recent cancer trends in the United States. J Natl Cancer Inst. 1995;87:175-82. [ Links ]
3. Isaacs H Jr. Tumors of the fetus and newborn. En: Livolsi VA, editor. Majors problems in pathology series, 35. Philadelphia WB Saunders, (1997). [ Links ]
4. Berry PJ. Congenital tumours. En: Keeling JW, editor. Fetal and neonatal pathology. Berlin: Springer-Verlag; 1993. pp. 273-294. [ Links ]
5. Bader JL, Miller RW. US cancer incidence and mortality in the first year of life. Am J Dis Child. 1979;133:157-9. [ Links ]
6. Vasilatou-Kosmidis H. Cancer in neonates and infants. Med Pediatr Oncol. 2003;41:7-9. [ Links ]
7. Ries LAG, Smith MA, Gurney JG, Linet M, Tamra T, Young JL, et al, eds. Cancer incidence and survival among children and adolescents: United States SEER Program 1975-2008 [consultado 23 May 2013]. Disponible en: http://seer.cancer.gov/publications/childhood/. [ Links ]
8. Vormoor J, Chintagumpala M. Leukaemia and cancer in neonates. Semin Fetal Neonatal Med. 2012;17:183-4. [ Links ]
9. Cicero-Oneto C, Zapata-Tarrés MM, Flores-Toscano KY, Flores-Montes OA. Tumores sólidos malignos neonatales en el Hospital Infantil de México Federico Gómez. Experiencia de 69 años. GAMO. 2013;12:143-9. [ Links ]
10. Magdum SA. Neonatal brain tumours-a review. Early Hum Dev. 2010;86:627-31. [ Links ]
11. Lakhoo K. Neonatal teratomas. Early Hum Dev. 2010;86:643-7. [ Links ]
12. Makin E, Davenport M. Fetal and neonatal liver tumours. Early Hum Dev. 2010;86:637-42. [ Links ]
13. Isaacs H Jr. Fetal and neonatal hepatic tumors. J Pediatr Surg. 2007;42:1797-803. [ Links ]
14. Isaacs H Jr. Fetal and neonatal renal tumors. J Pediatr Surg. 2008;43:1587-95. [ Links ]
15. Powis M. Neonatal renal tumours. Early Hum Dev. 2010;86:607-12. [ Links ]
16. Lakhoo K, Sowerbutts H. Neonatal tumours. Pediatr Surg Int. 2010;26:1159-68. [ Links ]
17. Couture A, Baud C, Veyrac C, Saguintaah M. Diagnostic imaging of neonatal tumors: Certainties and uncertainties. Arch Pediatr. 2001;8:278s-80s. [ Links ]
18. Donoghue V, Ryan S, Twomey E. Perinatal tumours: The contribution of radiology to management. Pediatr Radiol. 2008;38:S477-83. [ Links ]
19. Heaton TE, Liechty KW. Postnatal management of prenatally diagnosed abdominal masses and anomalies. Prenat Diagn. 2008;28:656-66. [ Links ]
20. Kawakami T, Monobe Y, Monforte H, Woods WG, Tuchman M, Lemieux B, et al. Pathology review of screening negative neuroblastomas: A report from the Quebec Neuroblastoma Screening Project. Cancer. 1998;83:575-81. [ Links ]
21. Cheung NK, Zhang J, Lu C, Parker M, Bahrami A, Tickoo SK, et al. Association of age at diagnosis and genetic mutations in patients with neuroblastoma. JAMA. 2012;307:1062-71. [ Links ]
22. Parker L, Powell J. Screening for neuroblastoma in infants younger than 1 year of age: Review of the first 30 years. Med Pediatr Oncol. 1998;31:455-69. [ Links ]
23. Nishihira H, Toyoda Y, Tanaka Y, Ijiri R, Aida N, Takeuchi M, et al. Natural course of neuroblastoma detected by mass screeing: A 5-year prospective study at a single institution. J Clin Oncol. 2000;18:3012-7. [ Links ]
24. Lee TC, Olutoye OO. Evaluation of the prenatally diagnosed mass. Semin Fetal Neonatal Med. 2012;17:185-91. [ Links ]
25. VanDorsten JP, Hulsey TC, Newman RB, Menard MK. Fetal anomaly detection by second-trimester ultrasonography in a tertiary center. Am J Obstet Gynecol. 1998;178:742-9. [ Links ]
26. Lazar DA, Olutoye OO, Moise KJ Jr, Ivey RT, Johnson A, Ayres N, et al. Ex-utero intrapartum treatment procedure for giant neck masses-fetal and maternal outcomes. J Pediatr Surg. 2011;46:817-22. [ Links ]
27. Degueldre SC, Chockalingam P, Mivelaz Y, di Bernardo S, Pfammatter JP, Barrea C, et al. Considerations for prenatal counselling of patients with cardiac rhabdomyomas based on their cardiac and neurologic outcomes. Cardiol Young. 2010;20:18-24. [ Links ]
28. Xu K, Rosenwaks Z, Beaverson K, Cholst I, Veeck L, Abramson DH. Preimplantation genetic diagnosis for retinoblastoma: The first reported liveborn. Am J Ophthalmol. 2004;137:18-23. [ Links ]
29. Dommering CJ, Moll AC, Imhof SM, de Die-Smulders CE, Coonen E. Another liveborn after preimplantation genetic diagnosis for retinoblastoma. Am J Ophthalmol. 2004;138:1088-9. [ Links ]
30. Chamberlain J. Screening for neuroblastoma: a review of the evidence. J Med Screen. 1994;1:169-75. [ Links ]
31. Challis GB, Stam HJ. The spontaneous regression of cancer. A review of cases from 1900 to 1987. Acta Oncol. 1990;29:545-50. [ Links ]
32. Ferrís i Tortajada J, García i Castell J, López-Andreu JA, Berbel-Tornero O. Factores ambientales asociados a cánceres pediátricos. Rev Esp Pediatr. 1999;55:166-77. [ Links ]
33. Satgé D, Sasco AJ, Little J. Antenatal therapeutic drug exposure and fetal/neonatal tumours: Review of 89 cases. Paediatr Perinat Epidemiol. 1998;12:84-117. [ Links ]
34. Satgé D, Sommelet D, Geneix A, Nishi M, Malet P, Vekemans M. A tumor profile in Down syndrome. Am J Med Genet. 1998;78:207-16. [ Links ]
35. Bolande RP. Prenatal carcinogenesis. An appraisal. Cancer. 1994;74:1674-9. [ Links ]
36. Berbel Tornero O, Ferrís i Tortajada J, Donat Colomer J, Ortega García JA, Muñoz Guillén A, Verdeguer Miralles A. Tumores neonatales: características clínicas y terapéuticas. Análisis de 72 casos del hospital infantil La Fe de Valencia. An Pediatr (Barc). 2006;65:108-17. [ Links ]
37. Berbel Tornero O, Ortega García JA, Ferrís i Tortajada J, García Castell J, Donat i Colomer J, Soldin OP, et al. Neonatal tumours and congenital malformations. An Pediatr (Barc). 2008;68:589-95. [ Links ]
38. Preston-Martin S. Epidemiological studies of perinatal carcinogenesis. IARC Sci Publ. 1989;289-314. [ Links ]
39. Bearer CF. Environmental health hazards: How children are different from adults. Future Child. 1995;5:11-26. [ Links ]
40. Tomatis L. Prenatal carcinogenesis. IARC Sci Publ. 1988;121-32. [ Links ]
41. Anderson LM, Jones AB, Rice JM. Perinatal carcinogenesis: Current directions. Br J Cancer. 1991;63:1025-8. [ Links ]
42. Alexandrov V, Aiello C, Rossi L. Modifying factors in prenatal carcinogenesis (review). In Vivo. 1990;4:327-35. [ Links ]
43. Benirschke K. The placenta in the litigation process. Am J Obstet Gynecol. 1990;162:1445-8. [ Links ]
44. Berbel-Tornero O, Ortega-Garcia JA, Ferris-Tortajada J. Congenital abnormalities and childhood cancer: A cohort record-linkage study. Cancer. 2006;106:1418-9. [ Links ]
45. Sadetzki S, Mandelzweig L. Childhood exposure to external ionising radiation and solid cancer risk. Br J Cancer. 2009;100:1021-5. [ Links ]
46. Inskip H. Stillbirth and paternal preconceptional radiation exposure. Lancet. 1999;354:1400-1. [ Links ]
47. Doll R, Wakeford R. Risk of childhood cancer from fetal irradiation. Br J Radiol. 1997;70:130-9. [ Links ]
48. Fattibene P, Mazzei F, Nuccetelli C, Risica S. Prenatal exposure to ionizing radiation: Sources, effects and regulatory aspects. Acta Paediatr. 1999;88:693-702. [ Links ]
49. Friedman DR, Hatch EE, Tarone R, Kaune WT, Kleinerman RA, Wacholder S, et al. Childhood exposure to magnetic fields: Residential area measurements compared to personal dosimetry. Epidemiology. 1996;7:151-5. [ Links ]
50. UK Childhood Cancer Study Investigators. Exposure to power-frequency magnetic fields and the risk of childhood cancer. Lancet. 1999;4:1925-31. [ Links ]
51. Infante-Rivard C, Deadman JE. Maternal occupational exposure to extremely low frequency magnetic fields during pregnancy and childhood leukemia. Epidemiology. 2003;14:437-41. [ Links ]
52. De Roos AJ, Teschke K, Savitz DA, Poole C, Grufferman S, Pollock BH, et al. Parental occupational exposures to electromagnetic fields and radiation and the incidence of neuroblastoma in offspring. Epidemiology. 2001;12:508-17. [ Links ]
53. Feychting M, Ahlbom A. Health effects of exposure to magnetic fields? Epidemiology, unknown mechanisms. Lakartidningen. 2001;14:5168. (Sueco). [ Links ]
54. Stillerman KP, Mattison DR, Giudice LC, Woodruff TJ. Environmental exposures and adverse pregnancy outcomes: A review of the science. Reprod Sci. 2008;15:631-50. [ Links ]
55. Ferrís Tortajada J, Ortega García JA, Marco Macián A, García Castell J. Environment and pediatric cancer. An Pediatr (Barc). 2004;61:42-50 (Español). [ Links ]
56. Ortega-García JA, Ferrís-Tortajada J, Torres-Cantero AM, Soldin OP, Torres EP, Fuster-Soler JL, et al. Full breastfeeding and paediatric cancer. J Paediatr Child Health. 2008;44:10-3. [ Links ]
57. Ortega-García JA, Martin M, López-Fernández MT, Fuster-Soler JL, Donat-Colomer J, López-Ibor B. Transgenerational tobacco smoke exposure and childhood cancer: An observational study. J Paediatr Child Health. 2010;46:291-5. [ Links ]
58. Yang Q, Olshan AF, Bondy ML, Shah NR, Pollock BH, Seeger RC, et al. Parental smoking and alcohol consumption and risk of neuroblastoma. Cancer Epidemiol Biomarkers Prev. 2000;9:967-72. [ Links ]
59. Schüz J, Kaletsch U, Kaatsch P, Meinert R, Michaelis J. Risk factors for pediatric tumors of the central nervous system: Results from a German population-based case-control study. Med Pediatr Oncol. 2001;36:274-82. [ Links ]
60. Boffetta P. Health effects of asbestos exposure in humans: A quantitative assessment. Med Lav. 1998;89:471-80. [ Links ]
61. Colt JS, Blair A. Parental occupational exposures and risk of childhood cancer. Environ Health Perspect. 1998;106:909-25. [ Links ]
62. Weitzman S, Grant R. Neonatal oncology: Diagnostic and therapeutic dilemmas. Semin Perinatol. 1997;21:102-11. [ Links ]
63. Aigrain Y, Philippe-Chomette P. Surgical management of malignant tumors in newborns. Arch Pediatr. 2001;8:281s-2s. Francés. [ Links ]
64. Zucker JM. Indications for medical treatment for tumors in newborns. Arch Pediatr. 2001;8:283s-6s (Francés). [ Links ]
65. Zapata M, Juárez E, Murguía T. Abordaje del neonato con sospecha de cáncer. En: Murguía T, Villanueva D, Lara G, editores. Neonatología. Esencia, arte y praxis. México D.F: McGraw Hill; 2011 pp. 349-358. [ Links ]
66. Frazier AL, Weldon C, Amatruda J. Fetal and neonatal germ cell tumors. Semin Fetal Neonatal Med. 2012;17:222-30. [ Links ]
67. Dhir S, Wheeler K. Neonatal neuroblastoma. Early Hum Dev. 2010;86:601-5. [ Links ]
68. Hwang SW, Su JM, Jea A. Diagnosis and management of brain and spinal cord tumors in the neonate. Semin Fetal Neonatal Med. 2012;17:202-6. [ Links ]
69. Isaacs H Jr. II. Perinatal brain tumors: A review of 250 cases. Pediatr Neurol. 2002;27:333-42. [ Links ]
70. Van der Linden MH, Creemers S, Pieters R. Diagnosis and management of neonatal leukemia. Semin Fetal Neonatal Med. 2012;17:192-5. [ Links ]
71. Roy A, Roberts I, Vyas P. Biology and management of transient abnormal myelopoiesis (TAM) in children with Down syndrome. Semin Fetal Neonatal Med. 2012;17:196-201. [ Links ]
72. Thompson PA, Chintagumpala M. Renal and hepatic tumors in the neonatal period. Semin Fetal Neonatal Med. 2012;17:216-21. [ Links ]
73. Bader D, Riskin A, Vafsi O, Tamir A, Peskin B, Israel N, et al. Alpha-fetoprotein in the early neonatal period-a large study and review of the literature. Clin Chim Acta. 2004;349:15-23. [ Links ]
74. Parulekar MV. Retinoblastoma-current treatment and future direction. Early Hum Dev. 2010;86:619-25. [ Links ]
75. Ferrari A, Orbach D, Sultan I, Casanova M, Bisogno G. Neonatal soft tissue sarcomas. Semin Fetal Neonatal Med. 2012;17:231-8. [ Links ]

